Lung cancer cellular model (A549)-valuable fuel in CRISPR Therapeutics and Biomedicine research
A549 is a model lung cancer cell line developed in 1972 by scientist Giard, through the removal and culturing of cancerous lung tissue in the explanted tumor of a 58-year-old Caucasian male. A549 cells are used in a variety of basic cell biology and biomedical studies involving lung cancer proliferation, corresponding inhibitors studies, tumor formation, and metastases. Potential features that attract the investigators are epithelial cells with squamous nature and responsible for the diffusion of substances, such as water and electrolytes, across the alveoli of the lungs. They can grow adherently, as a monolayer and able to synthesize lecithin and contain high levels of unsaturated fatty acids, which are important to maintain membrane phospholipids.
To date, a large number of human non-small cell lung cancer cell lines are being used for both basic research and drug discovery. A549 cells are well-characterized among the human lung carcinoma/alveolar cell lines as well as having KRAS mutation and wild type EGFR. However, A549 cells harbor oncogenic KRASG12S mutation as well as over 250 genetic mutations (COSMIC cell lines project), including some known oncogenes and tumor suppressors. K-RASG12S oncogene is solely responsible for their proliferation.
Key molecular studies performed with A549 cell as anti-tumor drug permeability and efficacy analysis, infection assays, respiratory immunotoxicity tests, cell senescence studies, and cytokine expression profiling. A549 cells used as a type II pulmonary epithelial cell model for drug metabolism and as a transfection host. These cells also utilized to study a variety of molecular characteristics for human tumors in culture.
Approximately 85% of lung cancer patients are with the non-small-cell lung cancer (NSCLC) histological subtype for which surgical resection or radical chemoradiotherapy offers the best prospect of cure. However, the 5-year survival rate for patients with advanced-stage lung cancer is less than 6%, with the majority of patients die from metastatic, drug resistance, and recurrence of diseases. Additionally, multi-mutated and multigenic characteristics of lung cancer give them a unique heterogeneity ability that could exist differently in different cancer-affected patients with the same cancer type. Due to lung cancer's genetic and phenotypic diversity, individualization of therapy is becoming a reality; thus, this tumor entity is likely to further trigger future precision medicine development.
Gene editing technologies are rapidly advancing as a realistic therapeutic option. The ability to strategically edit a patient’s genome can constitute a treatment revolution. Genome editing technologies have huge potential in lung cancer treatment including targeting oncogenes and tumor-suppressor genes, genes related to chemotherapy drug resistance, and genes related to therapies using targeted drugs and inhibitors to promote further preclinical research and the clinical treatment of lung cancer.
Application:
1. Research tools for clinical research and mechanistic approaches; drug sensitivity and resistance; gene and antisense therapy; pathology, markers, and prognostic indicators; chemoprevention strategies; multimodality therapies, etc.
2. Standard research tool to analyze a variety of molecular biology procedures, for example, cell senescence, cytokine induction, protein expression, apoptosis, and receptor-ligand interactions.
3. Research tool to analyze the efficacy of potential anti-cancer agents to devise better cancer treatments for malignancies, such as non-small cell lung cancer (NSCLC).
4. Research tools suitable for producing adenoviruses that can be used in clinical trials and analyzing adenoviral-based therapies and vaccine strategies.
5. Research tool to study the infectivity of viruses that cause asthma to develop better asthma treatments.
6. Research tool for epigenetic studies, mutations in epigenetic regulators, epigenetic therapy, etc.
5. Research tools for disease modeling, cell line-based disease model, cell-based xenografts modeling either by subcutaneous or orthotopic injection or implantation of cells into mice to study cancer metastasis, tumor microenvironment, drug response, etc.
CRISPR-U™ gene editing in A549 cell line
The CRISPR system is a precise genome editing technique, which can create gene knockout or knock-in genome manipulations through the substitution of a target genetic sequence with a desired donor sequence. A549 is a model human lung adenocarcinoma epithelial cell line, used for lung cancer study mainly cancer vaccine, virus production, drug discovery, disease mechanisms, initiation, progression, and therapeutics. Investigators use CRISPR technique to engineered A549 cells to study lung cancer hallmarks, drug resistance mechanism, cancer therapeutics, cell death research, signaling pathways, drug discovery, drug response, and cell therapy. CRISPR/Cas9 technology positively fuel the advancement of in vivo and in vitro gene editing in lung cancer and have an immense impact on translational therapeutics. Ubigene developed CRISPR-U™ for gene manipulation of the A549 cell line. Thereby, possible to achieve genome-edited cells with the utilization of the CRISPR/Cas9 system. Ubigene can customize the gene-editing in eukaryotic cells as well as can generate various genes modification in animal models.
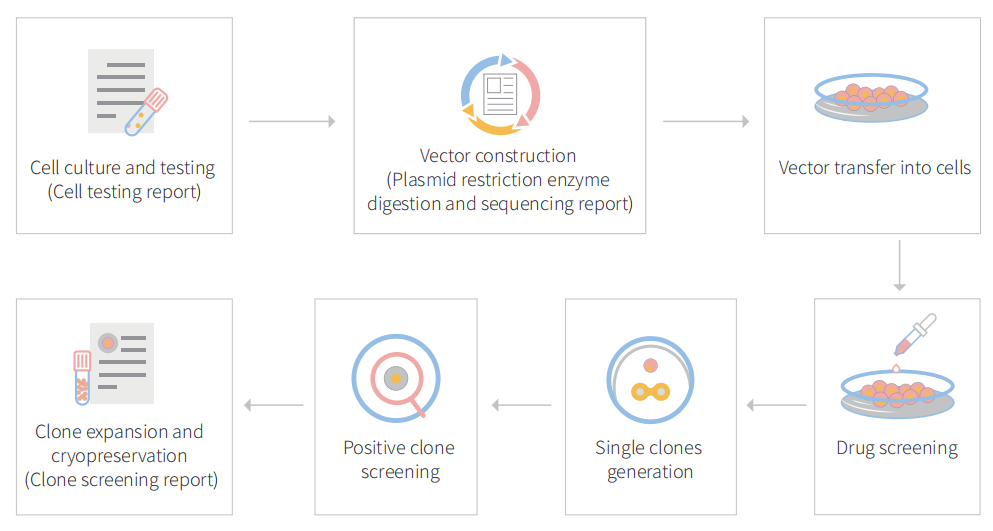
Figure: CRISPR-U™ customized workflow for engineered A549 model cells
Case study: Knock-out
NRF2 gene knockout restore the effectiveness of first-line chemotherapy against lung cancer
Drug resistance is a dilemma of lung cancer treatments. Resistance arises from the deregulation of various genes involved in controlling efflux or drug inactivation. In this study, the researcher reported the successful knockout of the NRF2 gene using CRISPR/Cas9 in chemo-resistant A549 lung cancer cells, with the subsequent demonstration of increased effectiveness of the anticancer drugs cisplatin, carboplatin, and vinorelbine in both cultures and a xenograft mouse model. The CRISPR designing and knockout of NRF2 experimental workflow shown in Fig.1
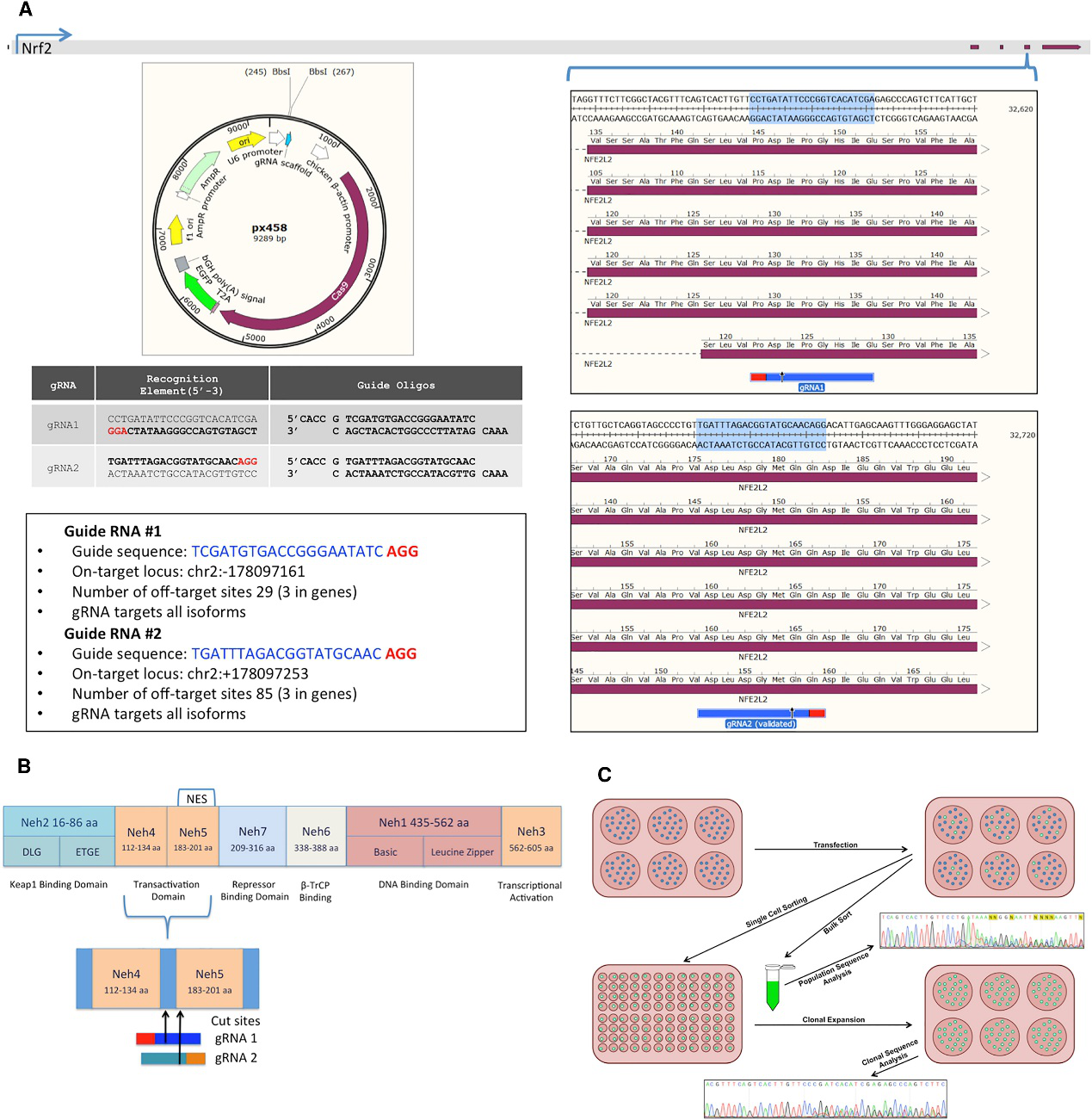
Fig 1: CRISPR Design and NRF2-Knockout Experimental Workflow
The cells were sorted based on GFP+ A549 cells transfected with either gRNA1 or gRNA2 and followed by sanger sequenced and analyzed for indel activity (Fig.2A). The clonally isolated NRF2-targeted cells were genomically analyzed for CRISPR/Cas9-induced NHEJ activity. Genomic DNA was Sanger sequenced and TIDE was used to develop the indel spectrums, sequence decompositions, and allelic patterns of NRF2, as shown for clones 1-40 and 2-11 (Fig.2B).
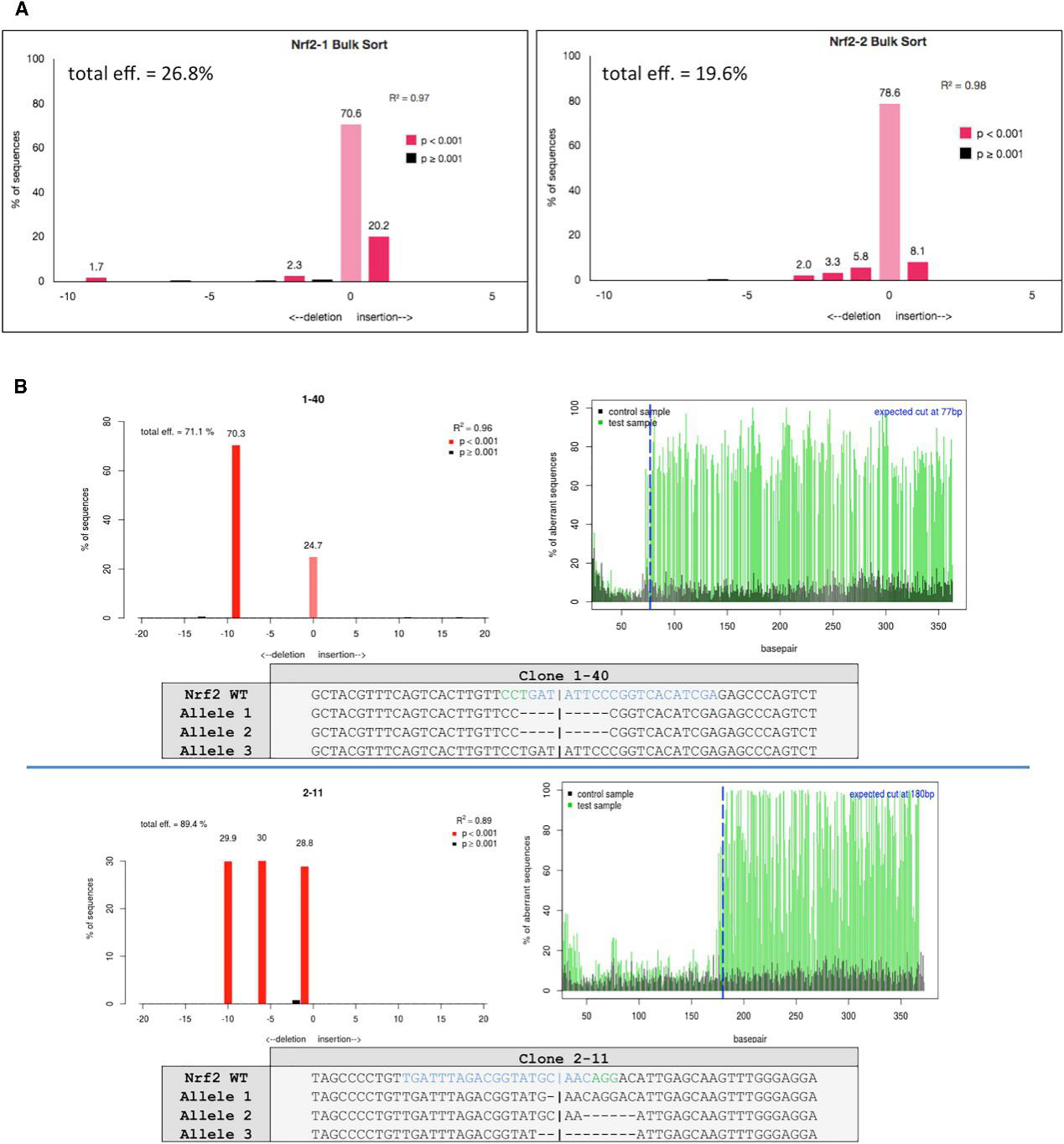
Fig 2: Genomic Analyses of NRF2-Knockout Clones
A fundamental cellular phenotype that could be affected by the lack of an NES is the rate at which cells proliferate in culture. They investigate the proliferation profile of clones 1-40 and 2-11 by staining the cells with antibodies against Ki67, followed by FACS analysis and western blot analysis (Fig. 3, A, B). Allelic analysis of clone 2-11 indicated that NRF2 is genetically disabled, and, when normalized to beta-actin and compared to wild-type A549 cells, clone 2-11 showed a knockdown of 68%.
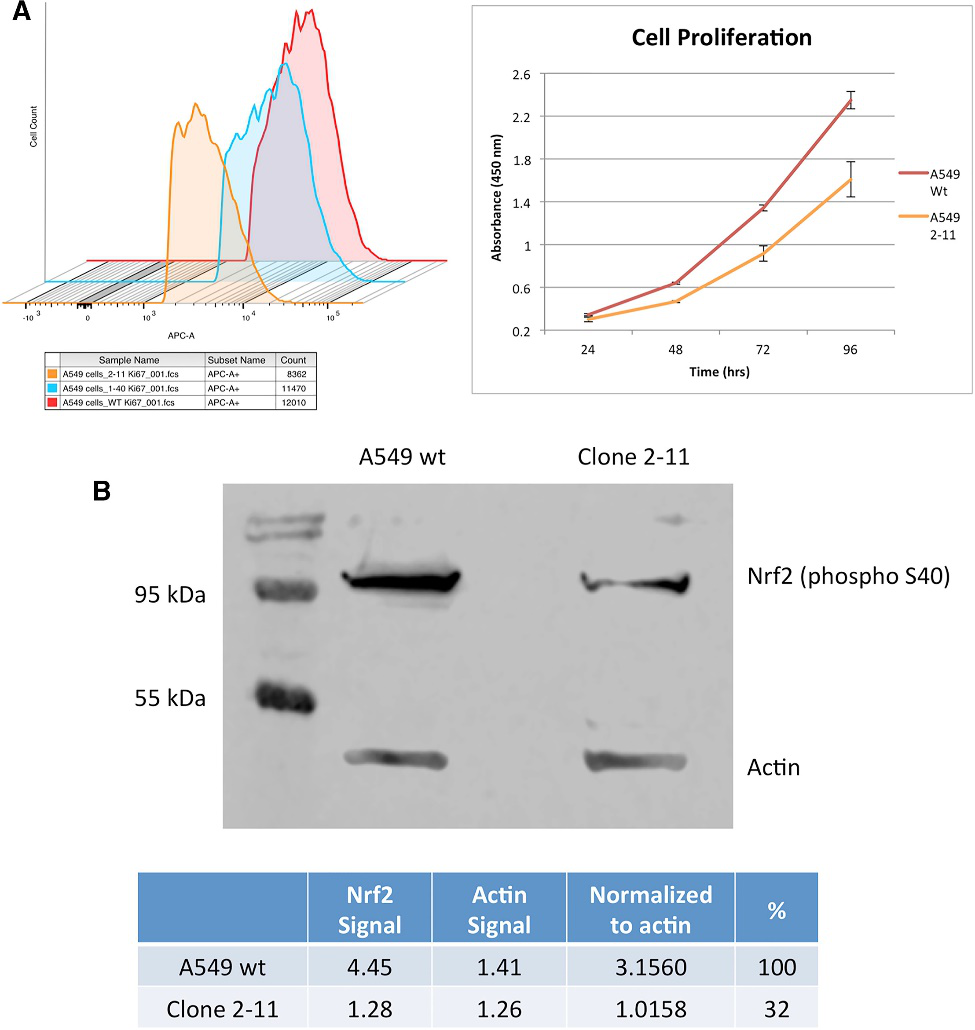
Fig 3: Cellular Proliferation Profile of NRF2- Knockout A549 Cells and Western Blot Analysis
They examine the chemosensitivity of the genetically engineered NRF2- deficient A549 cell lines, the MTS assay, depicted in Fig 4.
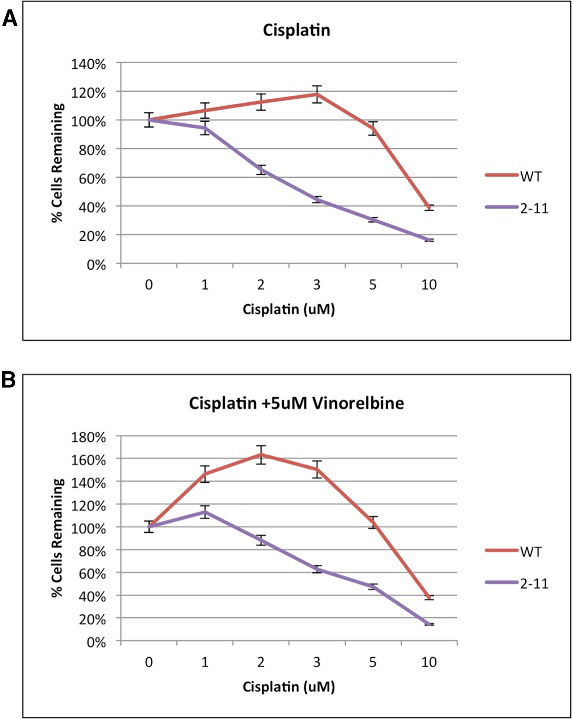
Fig 4: Proliferation Capacity of Wild-Type and NRF2-Modified A549 Cells (2-11) in Response to Chemotherapeutic Drugs
CRISPR/Cas9-mediated NRF2 knockout increased chemosensitivity in A549 cells in vitro and enhanced chemosensitivity driven by gene editing in a xenograft mouse model. The homozygous knockout A549 cells (clone 2-11) and wild-type A549 cells (control group) were implanted into the back of a nude mouse, and the cells (5 ×106 per cell line) were allowed to proliferate into a tumor with a diameter of approximately 100 mm3. The workflow is depicted in Figure 5A. Tumor growth through volume and proliferation was measured for 16 days, starting at the time of the first injection of the chemotherapeutic agent, day 0, and the results are presented in Fig. 5B–5D. A distinct difference was found among the four extracted tumor groups (Fig.5E).
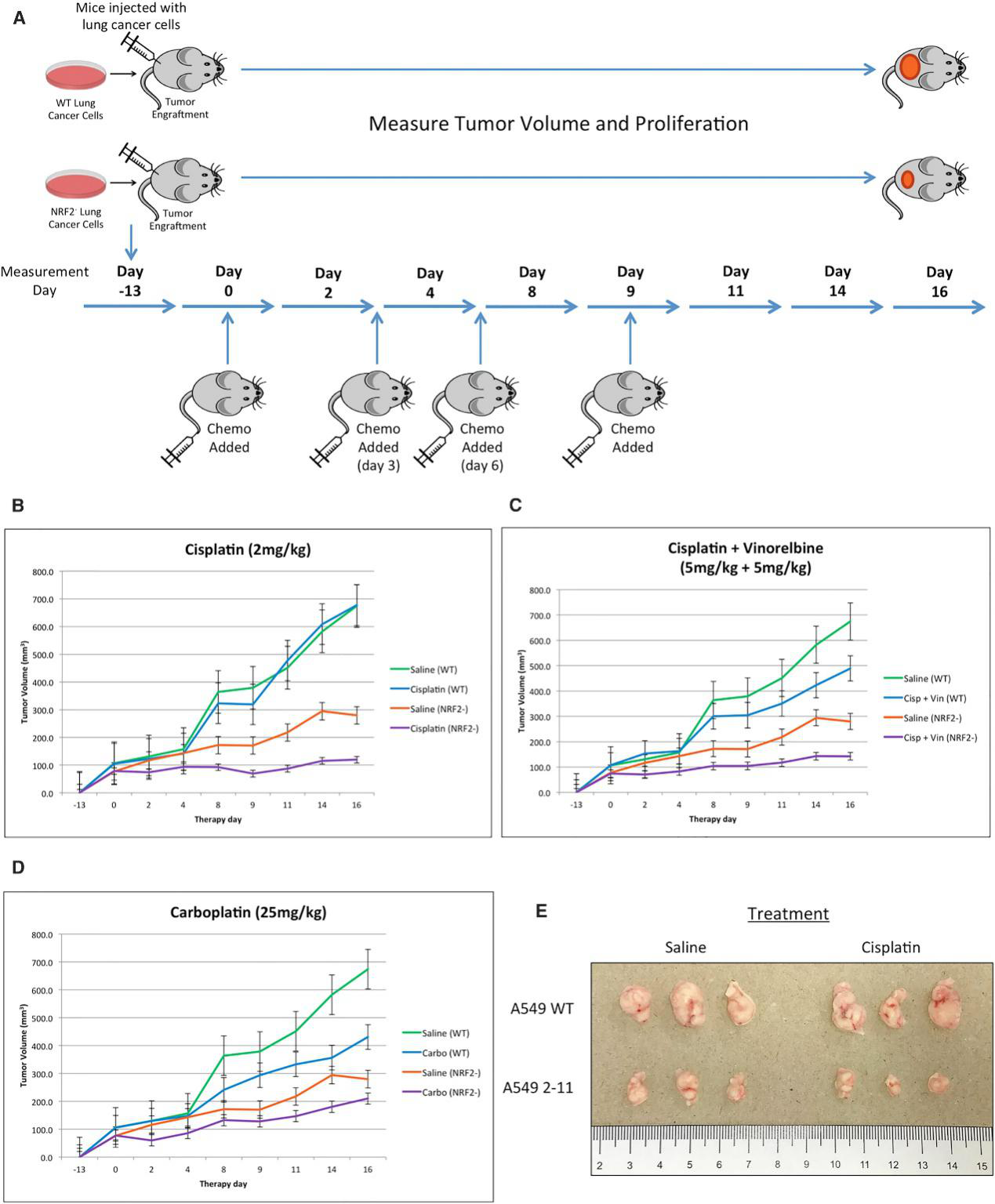
Fig 5: Restored Chemosensitivity in Mice with NRF2 Knockout in Tumors
Xenograft tumors extracted from mice, implanted with either wild-type A549 or NRF2-knockout A549 cells (2-11), 16 days after the initial treatment with either 2 mg/kg cisplatin or saline, were sectioned and stained with Ki67 (green) and DAPI (blue). Tumors generated from 2-11 cells shows noticeably decreased Ki67 staining, and treatment with cisplatin resulted in even lower levels of Ki67, suggesting cisplatin enhances the response of the knockout 2-11 cells by slowing down proliferation even further (Fig.6).
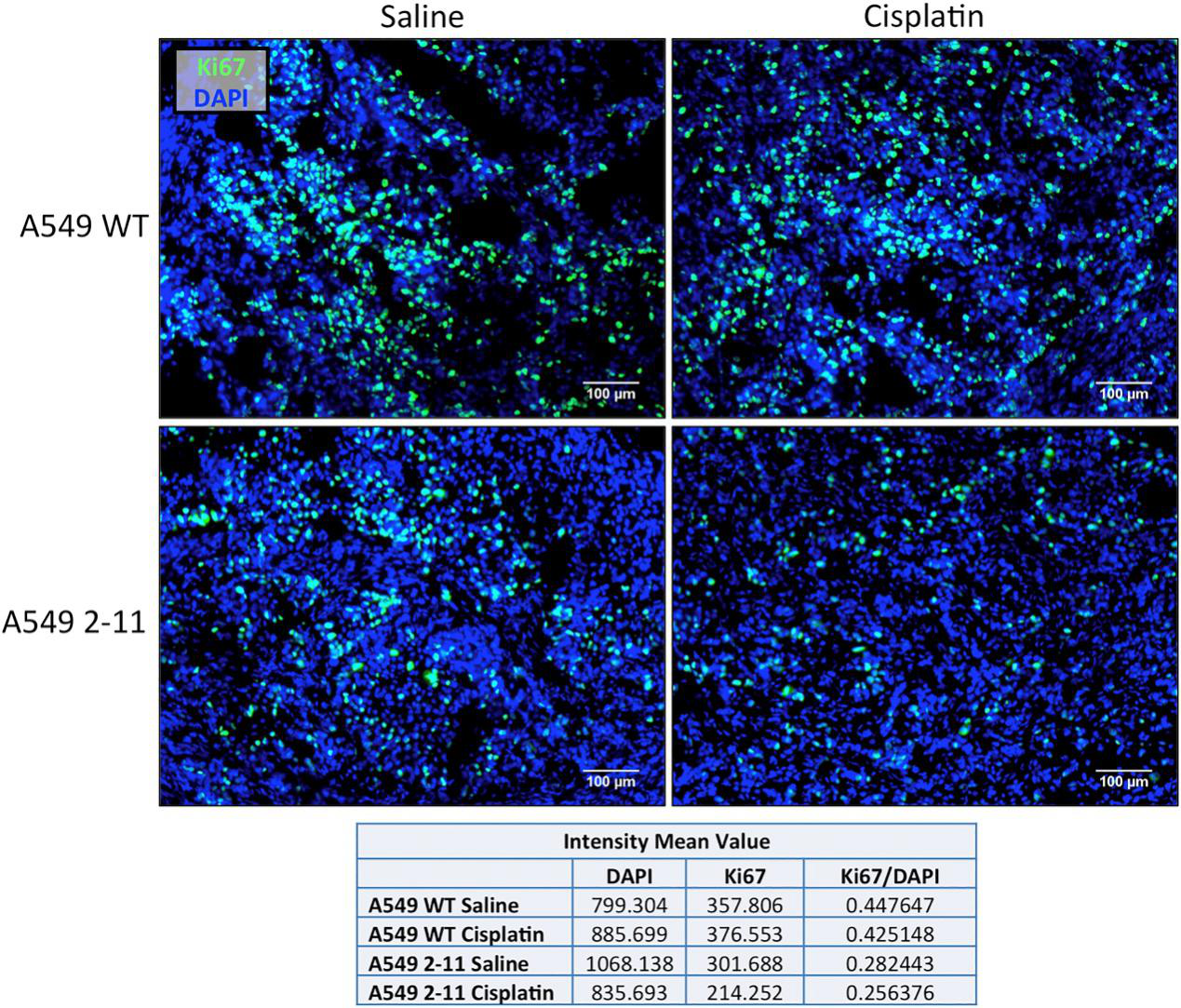
Fig 6: The proliferation of Xenograft Tumors
The effect of the disruption of the NES region located in the Neh5 domain of NRF2 was characterized using immunocytochemistry. Wild-type A549 and clone 2-11 cells were pre-treated with 2 mM cisplatin to stimulate NRF2 expression. Cells were quantified based on the following observed outcomes: no staining of NRF2, nuclear staining only, or cytoplasmic staining only. In wild-type cells, the majority of NRF2 was located in the nucleus, while in the functional knockout cell line (2-11), NRF2 was predominantly found in the cytoplasm (Fig.7)
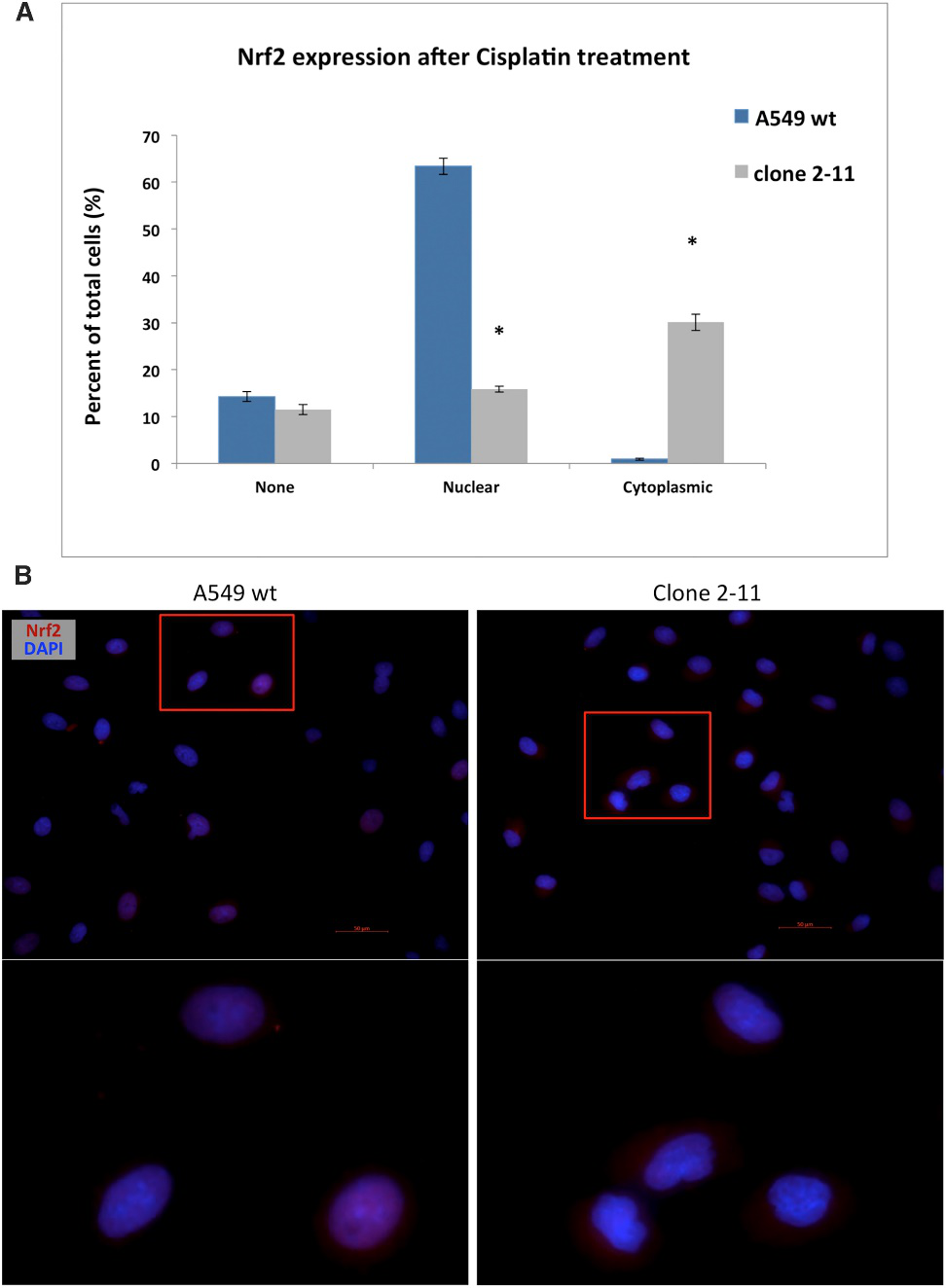
Fig 7: Cisplatin-Induced Nuclear and Cytoplasmic Localization of NRF2 in Wild-Type A549 Cells and Clone 2-11
In summary, the NRF2-knockout cells were less able to proliferate in culture and were more sensitive to chemotherapeutic agents including cisplatin and carboplatin. The NRF2-deficient cancer cells also grew more slowly than unmodified cancer cells when transplanted into mice. CRISPR-Cas9 gene editing can be used to restore the effectiveness of first-line chemotherapies against lung cancer, by knocking down a tumor gene that acts as a master regulator of genes involved in the development of resistance.
Case study: Knock-in
Targeted proteolysis of endogenous K-RAS is a viable strategy to target K-RAS-dependent pathologies
The oncogenicity of RAS mutations has been known for over three decades, intensive efforts have been made toward drugging them. There are three RAS oncogenes, H-RAS, K-RAS, and N-RAS, which represent the most frequently mutated genes in cancer and encode four highly similar proteins, namely H-RAS, N-RAS, K-RAS4A, and K-RAS4B. However, the development of conventional K-RAS inhibitors has been extremely challenging. Targeted proteolysis has emerged as a new modality in drug discovery to tackle undruggable targets. In this study, the researcher utilized targeted proteolysis using peptidic high-affinity binders, called AdPROM with CRISPR/Cas9 system to knock in a GFP tag on the native K-RAS gene in A549 adenocarcinoma (A549GFPKRAS) cells and constructed AdPROMs containing high-affinity GFP or H/KRAS binders. Expression of GFP-targeting AdPROM in A549GFPKRAS led to robust proteasomal degradation of endogenous GFP-K-RAS, while expression of anti-HRAS-targeting AdPROM in different cell lines resulted in the degradation of both GFP-tagged and untagged K-RAS, and untagged H-RAS.
CRISPR/Cas9 technology used to generate an A549 cell line harboring homozygous knock-in GFP protein cDNA at the N terminus of the native K-RAS gene (Fig.1). The cell's RAS protein expression was verified by western blot analysis using both panRAS and K-RAS4B antibodies as well as immunoprecipitation for GFP-K-RAS (Fig.1B, C). GFP-K-RAS localization study revealed that they are highly expressed in the plasma membrane, but not in mitochondria. A protein turnover validation was performed by using cycloheximide for WT K-RAS and GFP-K-RAS (Fig.1D, E).
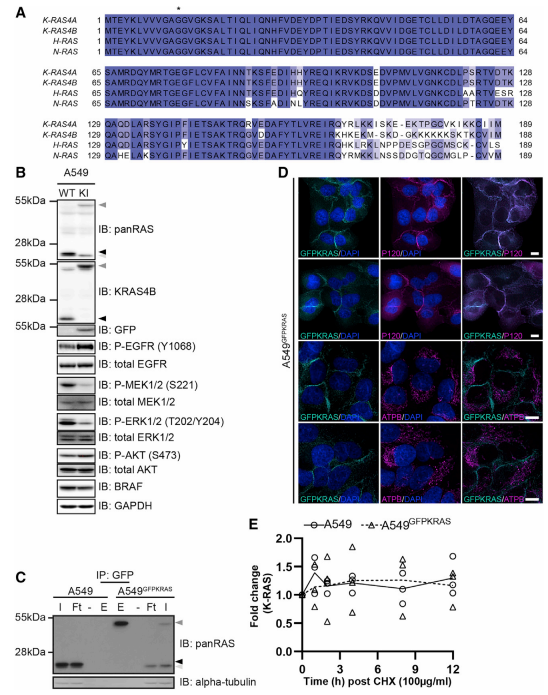
Fig 1: Generation of GFP-K-RAS Knockin in A549 NSCLC Cells by CRISPR/Cas9
Endogenously GFP-K-RAS protein is targeted for degradation by AdPROM system (Fig.2). A schematic representation of the proteolytic AdPROM system for endogenous GFP-K-RAS protein (Fig.2A). The expression of VHL-aGFP16 AdPROM resulting near-complete clearance of GFP-K-RAS from A549GFPKRAS cells (Fig.2B). The treatment of VHL-aGFP16 AdPROM expressing A549GFPKRAS cells with the Cullin neddylation inhibitor (MLN4924) partially rescued the degradation of GFP-K-RAS (Fig.2C). Around 98% of cells transduced with VHL-aGFP16 AdPROM virus showed GFP-K-RAS degradation as compared with untransduced A549GFPKRAS cells. A549GFPKRAS cells expressing VHL or aGFP16 alone were defined as GFP positive at 99.3% or 99.8%, respectively (Fig. 2D, E).
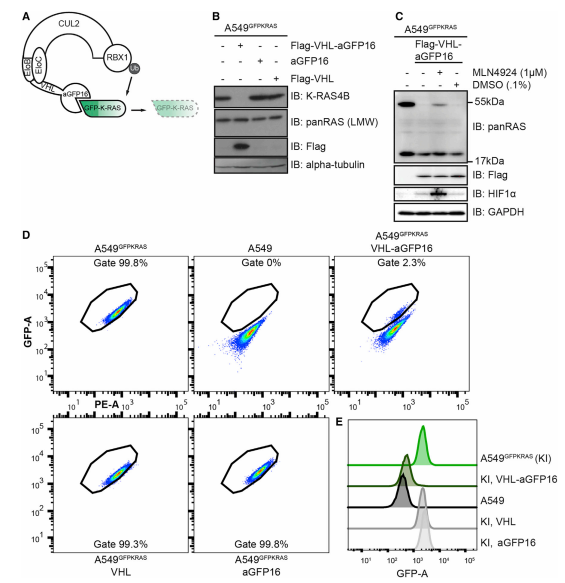
Fig 2: AdPROM-Mediated Degradation of GFP-K-RAS
To explore whether the AdPROM system can degrade endogenous, unmodified K-RAS from A549 cells, the researcher used a monobody anti-HRAS (aHRAS) with a FLAG tag to performed immunoprecipitates (IPs) experiments (Fig.3A). The degradation induced by VHL-aHRAS AdPROM was slightly less efficient than that achieved with the VHL-aGFP16 AdPROM (Fig.3B). The loss in protein levels of endogenous H-RAS protein caused by VHL-aHRAS AdPROM could be rescued by the Cullin neddylation inhibitor (MLN4924) (Fig.3C). Around 77% of cells showed degradation of GFP-K-RAS, as assessed by the shift of the GFP-positive gated population (Fig3. D, E). Uneven retroviral transduction of cells, which may account for the apparent uneven degradation of GFP-K-RAS through VHL-aHRAS (Fig.3F).
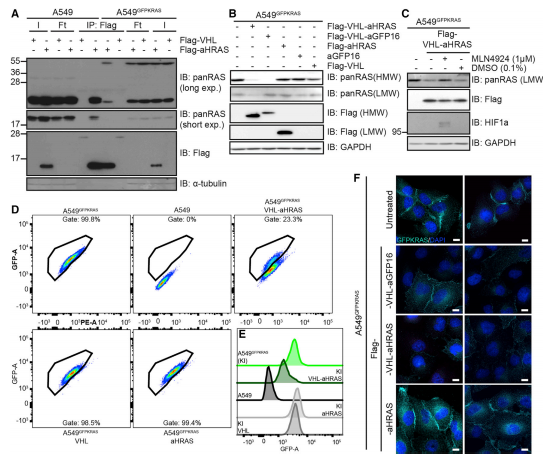
Fig 3: Degradation of Endogenous RAS Using a RAS-Specific Monobody
The ability to degrade endogenous untagged K- and H-/N-RAS in WT A549 cells were verified and they found that VHL-aHRAS resulted in a substantial reduction in apparent levels of both K-RAS (upper band) and H-/N-RAS (lower band) proteins as detected by the panRAS antibody (Fig.4A). They confirm the degradation occurs via the proteasome by cell treated with proteasomal inhibitors MG132 and bortezomib, both of which resulted in a strong accumulation of polyubiquitinated proteins (Fig.4B). They also found that RAS protein degradation slightly increases in K-RAS4A transcripts (Fig.4C) and performed global quantitative proteomic changes upon targeted degradation of RAS proteins (Fig4.D).
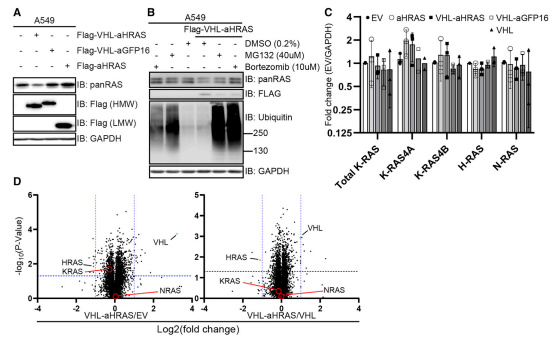
Fig 4:Degradation of Endogenous Unmodified RAS Using a RAS-Specific Monobody
The targeted degradation of K- and H-RAS proteins were also applied for WT A549, HT29, and SW620 cells (Fig.5)
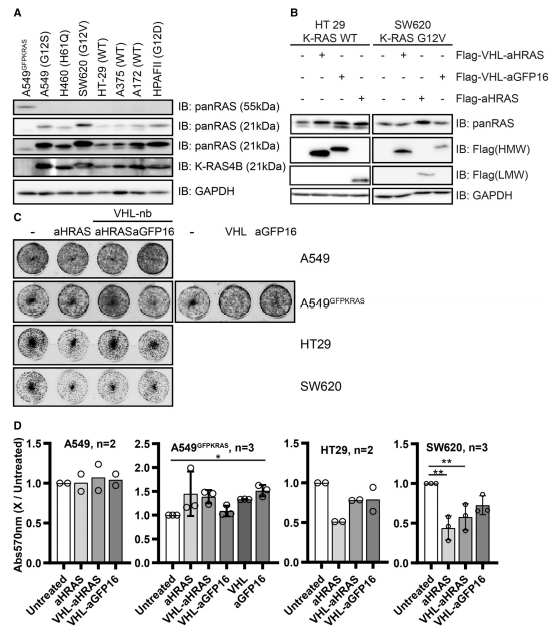
Fig 5:Degradation of RAS in Different Cell Lines and Effects on Proliferation
In summary, targeted proteolysis of endogenous K-RAS is a viable strategy to target K-RAS-dependent pathologies. The findings open up exciting opportunities to develop VHL-recruiting K-RAS-specific cell-permeable PROTACs as potential therapeutic agents. These findings also highlight the need for developing better and more selective RAS binding polypeptides, such as nanobodies or monobodies, to achieve more selective protein degradation.
Case study: Point mutation
EGFR mutation in KRAS mutated cells enhances TKI sensitivity
Driver mutations such as KRAS, ALK, and PI3K mutations co-exist with EGFR-mutations in a certain percentage of lung cancers. The therapeutic responses of such cases with co-mutations of driver genes are largely unclear. The investigators aimed to assess the response to EGFR-TKIs of cancer cells with EGFR/KRAS co-mutations.
CRISPR/Cas9-mediated genome editing applied to generated isogenic cell lines harboring one or two copies of an EGFR-activating mutation from A549 cell line, which is known to harbor a homozygous KRAS gene mutation. Human EGFR exon 21 sequences with ssODN repair template (Leu or Arg 858) are presented in Fig.1A. An overview of the successful workflow of transfection, HDR validated, and clonally expansion of L858R-knockin clones shown in Fig.1B.
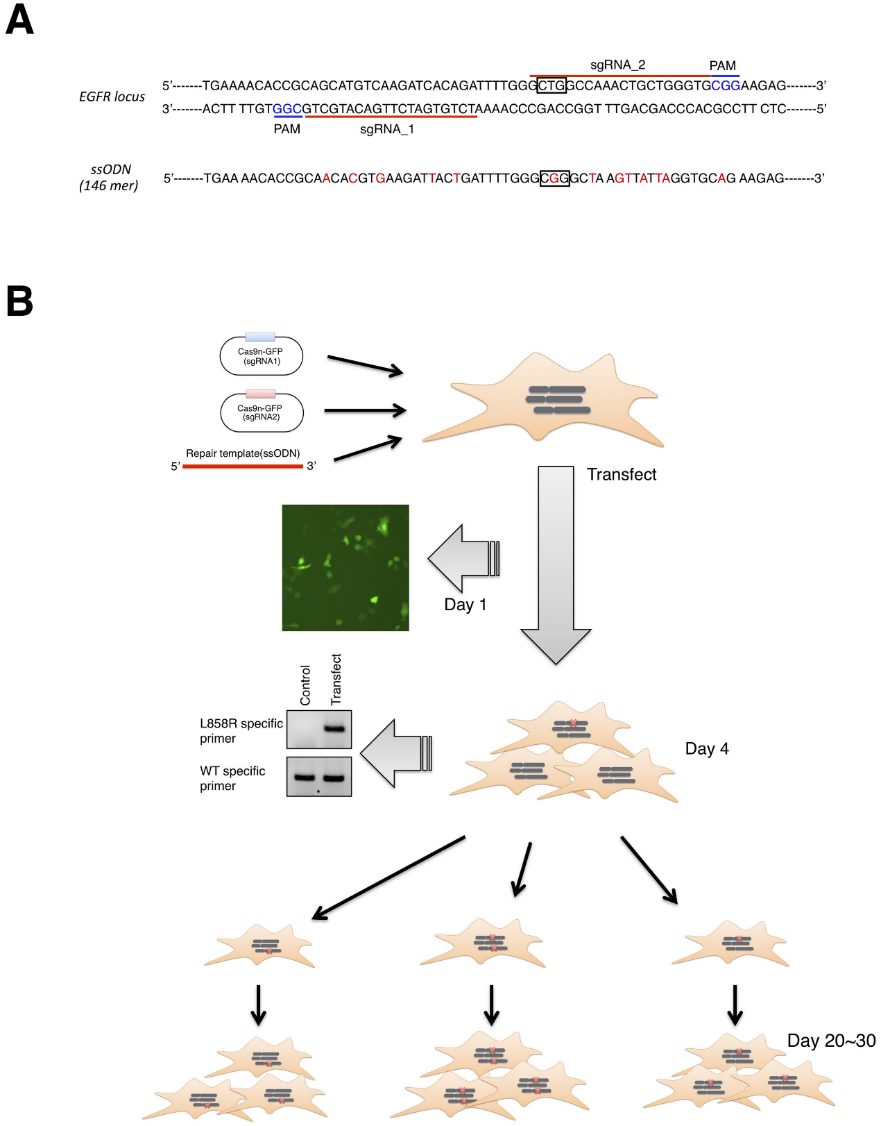
Fig 1: Overview of CRISPR/Cas9 genome editing
To confirm successful integration of the L858R EGFR mutation, the targeted genomic region was PCR-amplified and subcloned into a plasmid vector, and 20–30 bacterial colonies were sequenced for each clone and analyzed the sequences, mutation ratio, and copy number, etc (Fig.2).
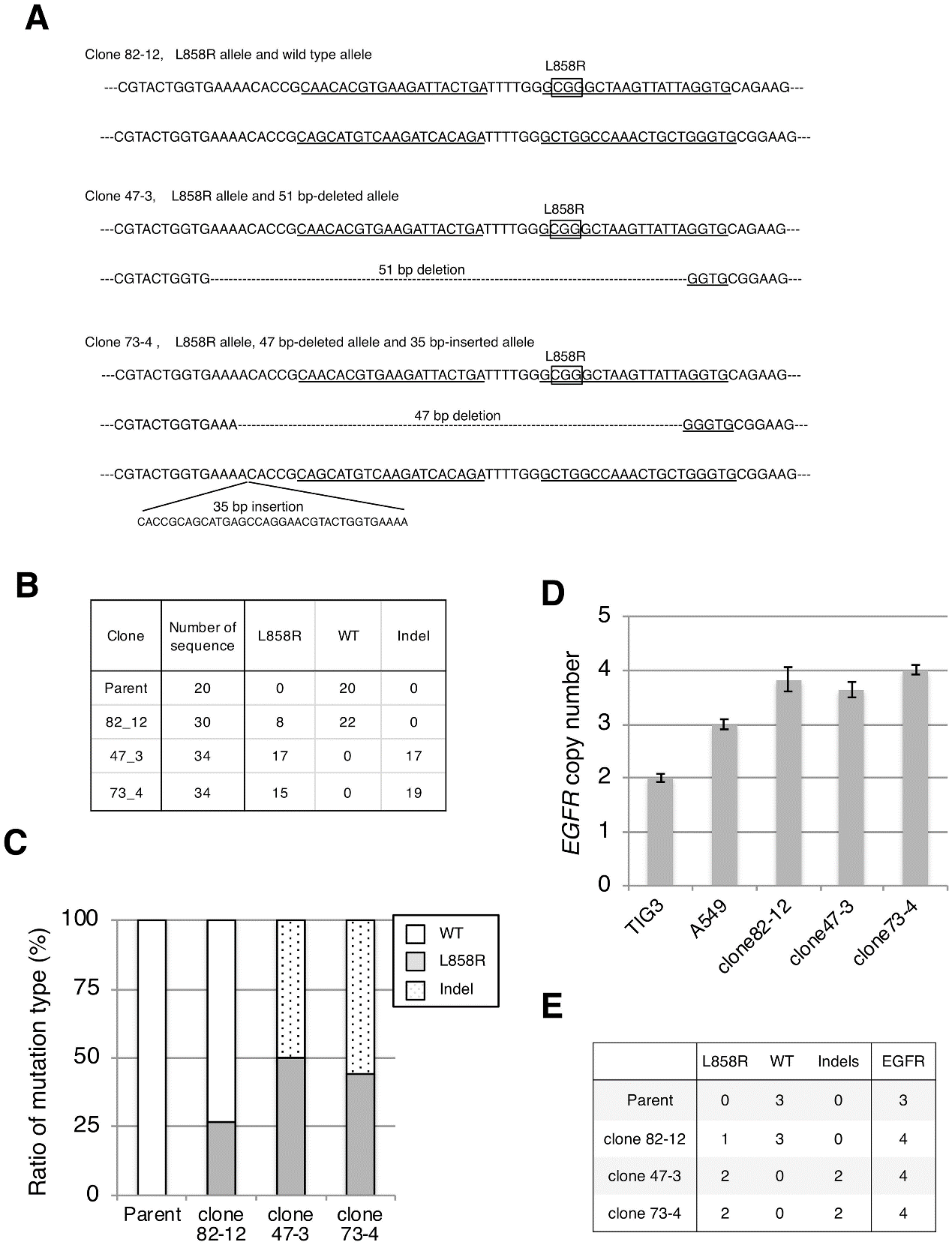
Fig 2: Establishment of cell lines harboring the L858R EGFR mutation.
The investigator analyses the expression of the L858R EGFR mutant in established clones at both the mRNA (Fig.3A, B) and protein levels (Fig.3C, D). The protein level of L858R EGFR in each clone correlated well with L858R EGFR mRNA expression (Fig.3).
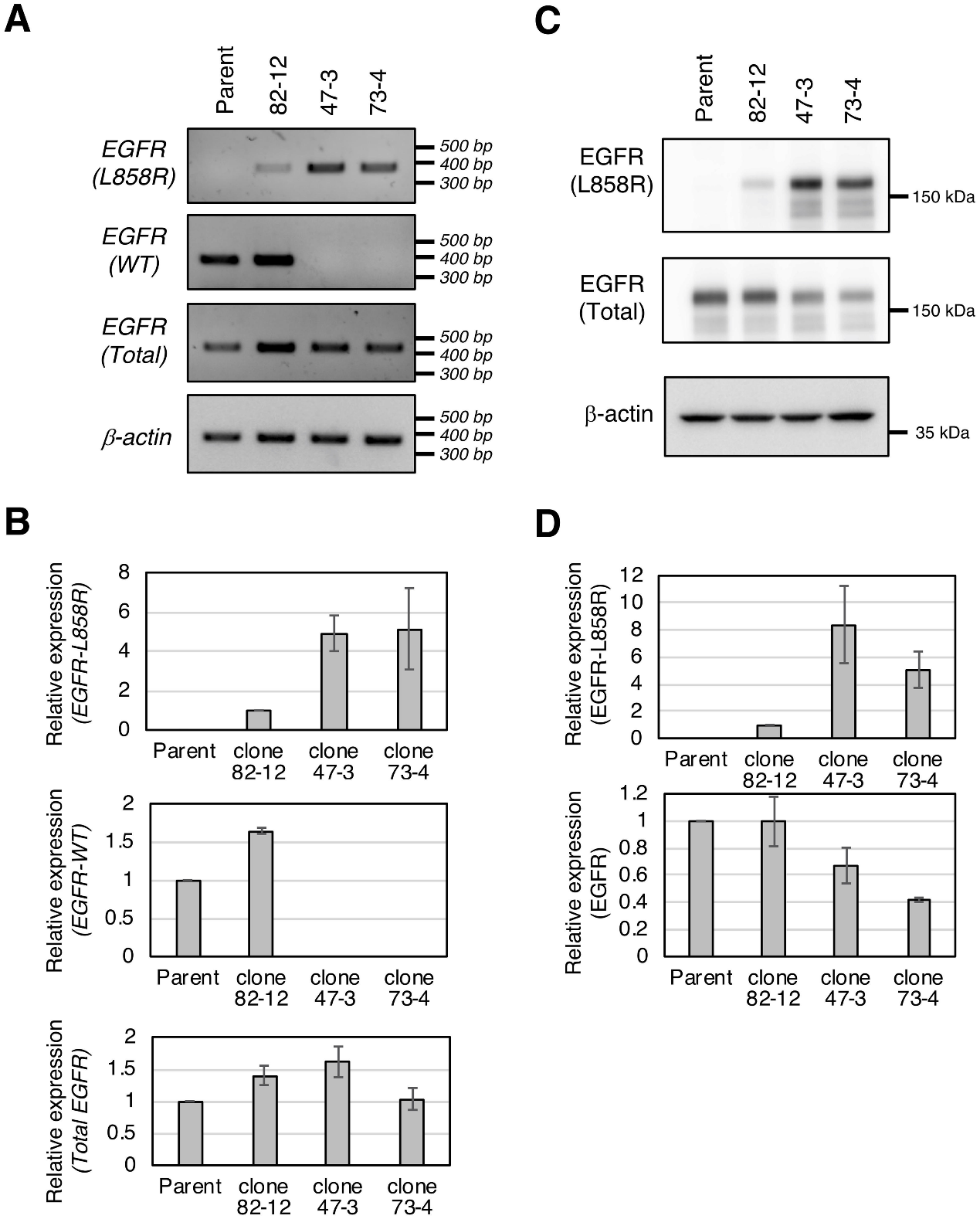
Fig 3: Expression of the L858R EGFR mutant in established clones.
The cells were treated with standard treatment options in NSCLC anti-cancer drug EGFR-TKIs (gefitinib and afatinib) and conventional anticancer drugs (cisplatin and taxol). The investigator found that all three clones were more sensitive to EGFR-TKIs than parental cells (Fig 4A). They also found that parental and clonal cells exhibit similar sensitivity to conventional anti-cancer drugs cisplatin and taxol (Fig.4B, C). Besides, clones 47–3 and 73–4 with two copies of the L858R mutation showed approximately 2.5- and 7-fold higher sensitivities to gefitinib and 25- and 10-fold higher sensitivities to afatinib than clone 82–12 with only one copy of L858R.
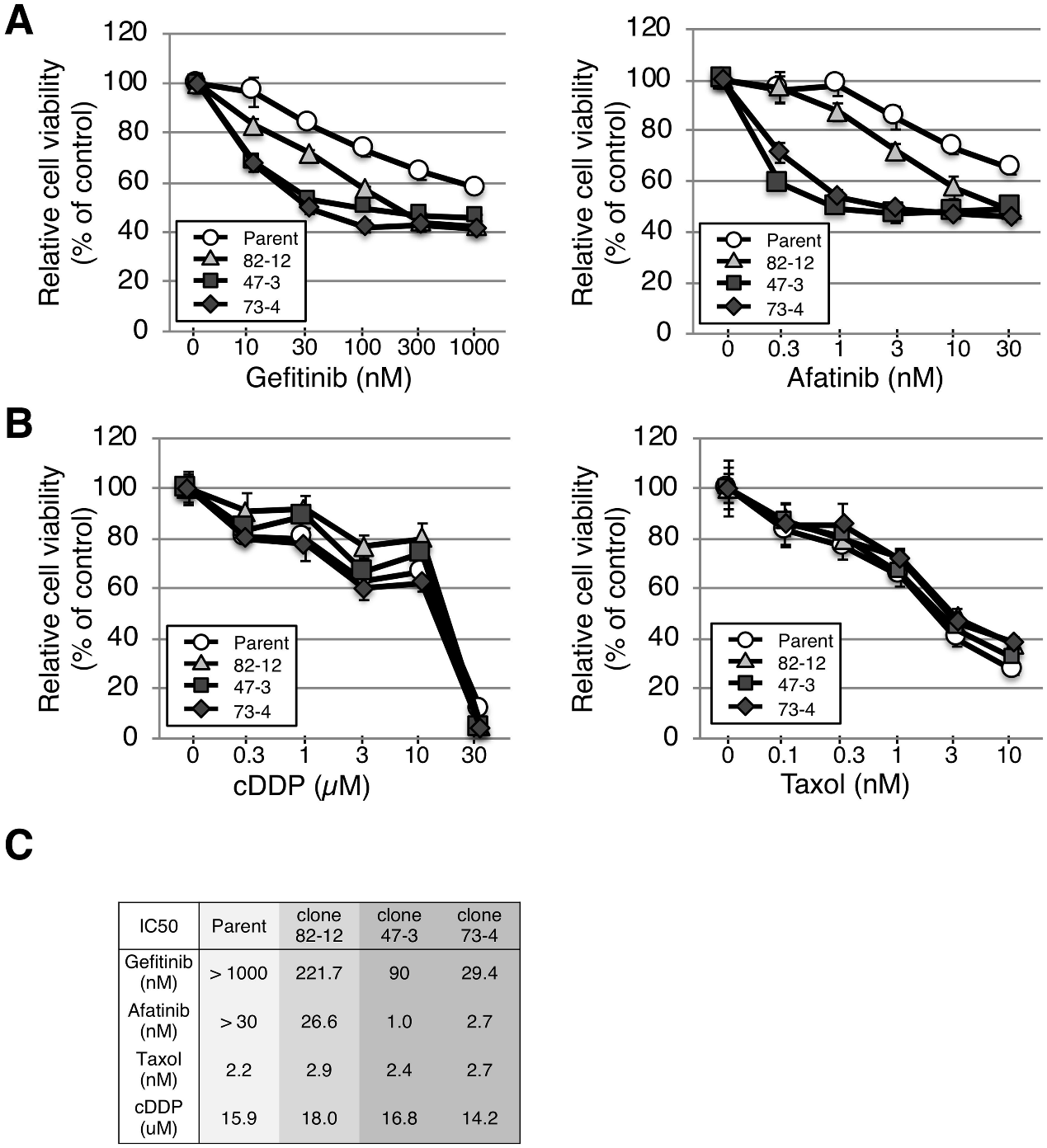
Fig 4:L858R EGFR mutation renders A549 cells sensitive to EGFR-TKIs.
Further investigation was carried out for the growth of each clone by the clonogenic survival assay in the presence of low-dose gefitinib (10 nM) and afatinib (0.3 nM) treatment. Strong growth inhibition was observed in gefitinib-treated clones with two copies of the L858R mutation (Fig 5A and 5B). Thus, clones with two copies of the L858R mutation were hypersensitive to TKIs compared with the clone with only one copy of the L858R mutation.
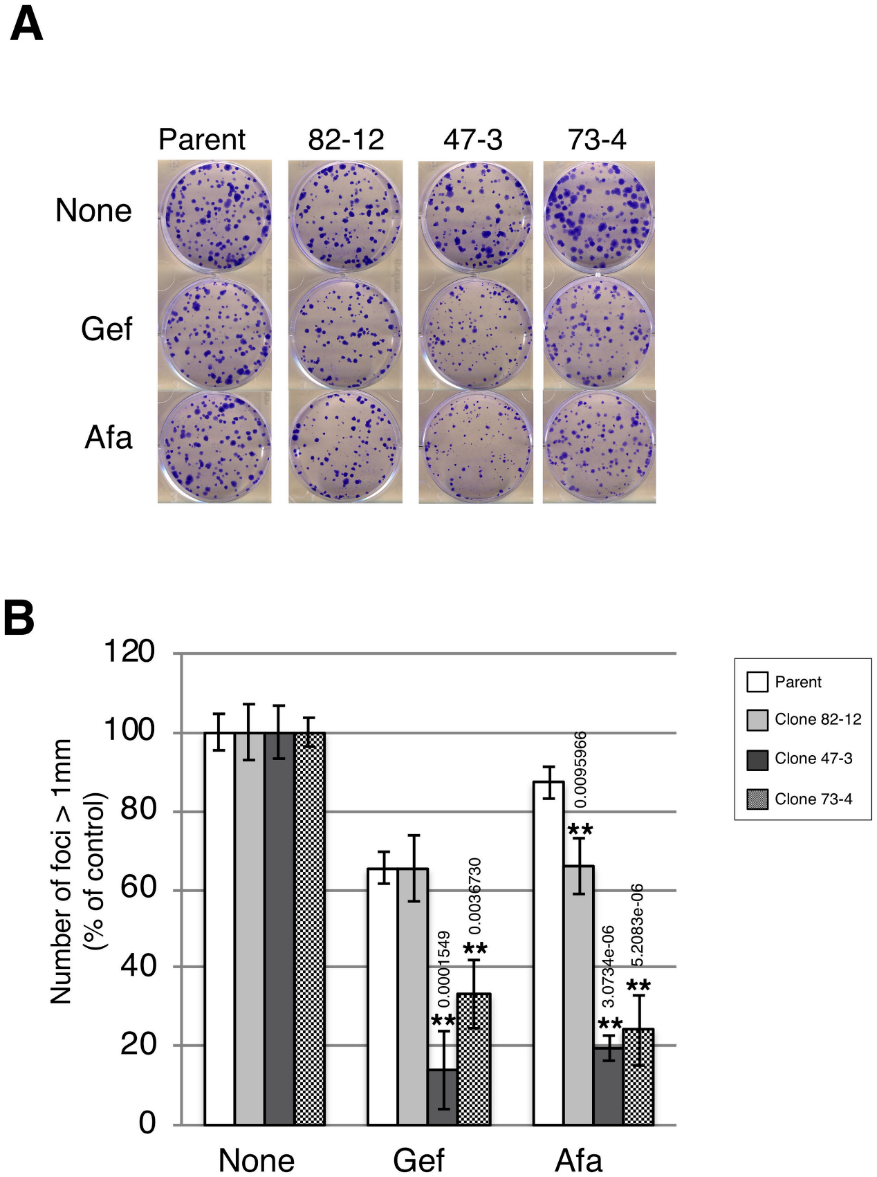
Fig 5:Two copies of L858R EGFR hypersensitize A549 cells to EGFR-TKIs
They investigated the EGFR phosphorylation status in each clone in the presence of gefitinib or afatinib. Gefitinib treatment decreased EGFR phosphorylation in a dose-dependent manner in each cell (Fig 6A and 6B). EGFR phosphorylation in clones 47–3 and 73–4, but not in parental cells or clone 82–12, was significantly reduced with afatinib treatment (p <0.01) (Fig. 6C, D). Thus, EGFR phosphorylation in clones 47–3 and 73–4 harboring two copies of the L858R mutation was markedly inhibited with lower doses of TKIs.
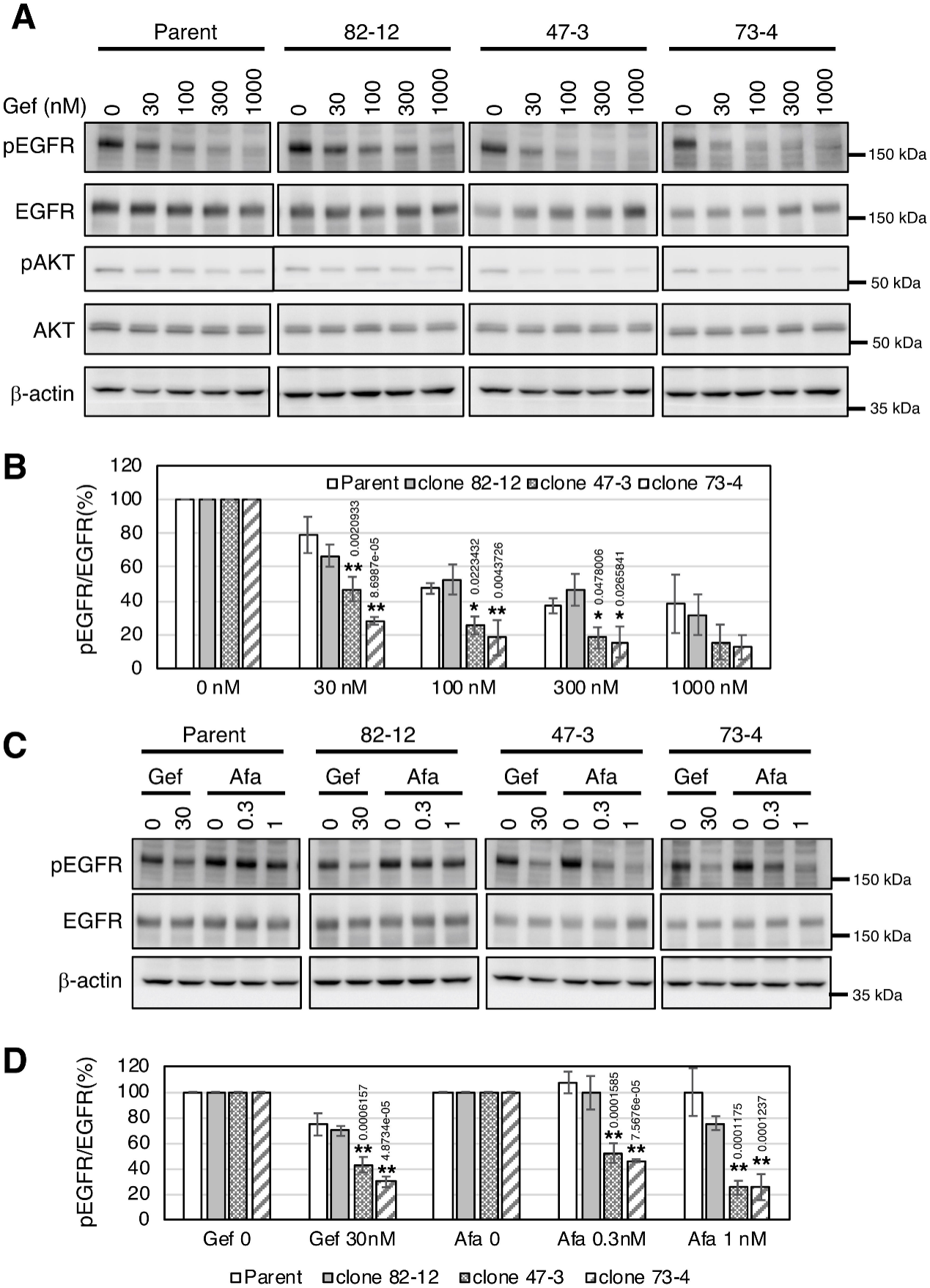
Fig 6:EGFR phosphorylation is markedly reduced by TKI treatment in cells with two copies of L858R EGFR.
In summary, the coexistence of EGFR mutation in KRAS mutated cells enhances TKI sensitivity, especially in the presence of a higher EGFR mutation copy number. Given that no effective targeted therapy against KRAS-mutated NSCLC is currently available, the identification and quantification of druggable mutations occurring concomitantly in tumors may provide an opportunity for the development of alternative treatment options for affected patients.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Reference:
Functional Gene Knockout of NRF2 Increases Chemosensitivity of Human Lung Cancer A549 Cells In Vitro and in a Xenograft Mouse Model. Mol Ther Oncolytics,2018,18;11:75-89.
Targeting Endogenous K-RAS for Degradation through the Affinity-Directed Protein Missile System. Cell Chem Biol, 2020, 27(9), 1151–1163.
Influence of EGFR-activating mutations on sensitivity to tyrosine kinase inhibitors in a KRAS mutant non-small cell lung cancer cell line, Plos One,2020,15(3):e0229712.



