Human CRISPR Knockout Pooled Library (Brunello)(Human CRISPR Knockout Pooled Library (Brunello))
Catalog#: LIBR-H015-LV1000;LIBR-H015-LV5000;LIBR-H015-LV10000
Size: 1*10^8TU
Instruction:

A detailed introduction of CRISPR library——the most popular high-throughput screening tool

The CRISPR library is the most popular high-throughput screening tool. In this article, Ubigene will give you a brief introduction of this high-throughput screening method from flowing aspects: what the CRISPR library is, what its applications are, classification introduction, selection strategy, construction process and use of CRISPR library. Hope this can provide you with some research ideas.
In this article
CRISPR library is a high-throughput gene screening method based on CRISPR/Cas9 technology. Construct a plasmid library by high-throughput synthesis of sgRNA, package it as lentivirus, and then infect the cells with low MOI (usually < 0.3), to ensure that only one sgRNA enters a cell, and then screen the cells (such as drug treatment, hypoxia treatment, virus infection or the self viability of the cells), collect the screened cells for NGS sequencing, and analyze the enrichment or depletion of sgRNA between the experimental group and the control group, then obtain host factors related to the screening condition.
Compared with cDNA library and RNAi library, CRISPR/Cas9 has the advantages of versatility, low noise, high knockout efficiency, and less off-target effect, and is the preferred method for large-scale gene function screening.
In addition to the unique advantages mentioned above, the broad application prospect also contributes to the popularity of CRISPR library. Here are a few common applications.
2.1.1 Identify essential genes of tumor and target oncogenes for treatment
Malignant tumor accumulates many gene mutations, but the maintenance of malignant phenotype of these tumor cells usually depends on only one or some of the oncogenes activated by mutations. This phenomenon is called oncogene addiction, and these oncogenes are also called driver oncogenes. Blocking the activity of these driver oncogenes can induce tumor cells to rapid apoptosis, growth arrest or differentiation, but with little impact on normal cells. This difference brings hope for targeted therapy of tumors.
In recent years, the extensive application of second-generation sequencing has identified many mutation loci in tumor cells, but it is difficult to distinguish which are driver mutations. It is a simpler and more direct method to find out the driving oncogenes that tumor cells depend on for survival by gene inactivation. CRISPR library can simultaneously target different genes throughout the genome, which is an excellent tool to achieve high-throughput gene inactivation. In 2014, Shalem et al. [1] first identified the key genes for the survival of human melanoma cells and pluripotent stem cells using the GeCKO Library (Fig. 1). Wang et al. [2] identified NCAPG as an essential oncogene for hepatocellular carcinoma tumor growth by CRISPR whole genome library screening. Kiessling et al. [3] verified the driving genes EGFR of HCC-827 cell line and NRAS and MEK1 of CHP-212 cell line by using CRISPR whole genome library screening, and found new driving genes TBK1 and TRIB2 for further research.
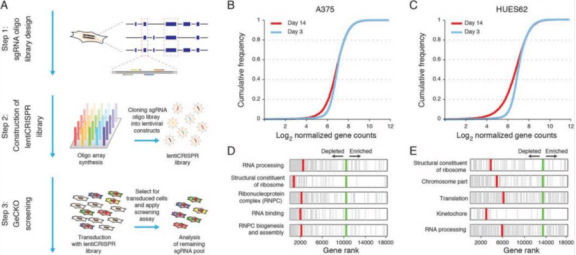
Fig. 1 Screening A375 and HUES62 survival essential genes by CRISPR library
2.1.2 Screen synthetic lethality genes to achieve precise treatment
For oncogenes, inhibitors can be used for targeted therapy, but for tumor suppressor genes, the inactivation of their functions makes it impossible to directly target them. So the synthetic lethality gene of the suppressor gene is usually targeted and inactivated. Synthetic lethality refers to the phenomenon that two non-lethal mutant genes inactivate at the same time leading to cell death. If there is inactivation of gene A in tumor cells, drugs can be used to inhibit its synthetic lethality gene B. By inactivate both genes in tumor cells, cell death would occur. However, healthy somatic cells can still have normal physiological functions because they have normal gene A. Thus, the drug specifically kills tumor cells (see Fig. 2) [4].
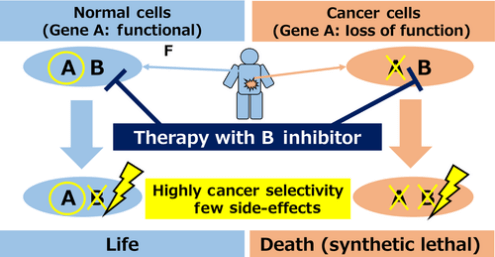
Fig.2 Synthetic lethality therapy
The most typical application of synthetic lethality is the treatment of BRCA1/2 mutated tumors with PARP inhibitors. By 2020, the global sales of PARP inhibitors had exceeded 2 billion US dollars. So how to find more gene pairs with synthetic lethality? CRISPR library high-throughput screening plays a big role! CRISPR library screening for synthetic lethality generally has two logics. One is to directly design a dual-gRNA vector, that is, to simultaneously target two genes in one cell and screen gene pairs with synthetic lethality. Shen et al. [5] used this method to screen 73 genes in three cancer cell lines (Hela, A549 and 293T) by 150000 combinations and found 120 gene pairs with synthetic lethality (see Fig. 3). Another logic is to screen a library on a single tumor suppressor gene knockout cell line to screen out targets that have synthetic lethal effects with the knockout gene. Feng et al.[6] used this method to first construct 12 known tumor suppressor gene knockout cell lines on 293A, and then screen wild-type and knockout cell lines with a genome-wide knockout library to analyze the changes in gRNA abundance before and after cell screening. Potential synthetic lethal genes were identified by comparing the abundance differences of gRNAs between wild-type and knockout cell lines after CRISPR library screening.
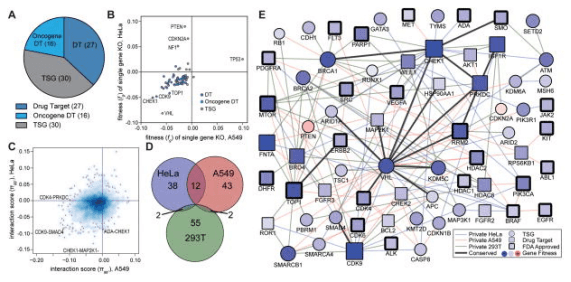
Fig. 3 Screening gene interaction relationships using CRISPR library
2.1.3 Search for tumor drug resistance related genes, study drug resistance mechanisms, and optimize treatment strategies
The main problem of tumor molecular targeted drugs is drug resistance, so it is of great clinical significance to explore the mechanism of drug resistance. Through positive screening of CRISPR library, most cells died due to drug sensitivity, and some cells survived due to drug resistance by knockout specific gene. The genes related to drug resistance can be screened out by detecting the gRNAs carried by the surviving cells.
Vemurafenib is approved for the treatment of advanced melanoma carrying the BRAFV600E mutation. In order to study the drug resistance mechanism of Vemurafenib, Shalem et al. [1] designed a knockout library containing 64751 sgRNAs and targeting 18080 genes in the whole genome. After cell infection and expansion, nf1, med12, nf2, cul3, tada2b, tada1 and other drug resistance related genes were screened out. A new hypothesis of the mechanism of Vemurafenib tumor resistance was proposed.
CRISPR library screening is often used to study host factors related to viral infection. For hepatitis C virus, dengue virus, Japanese encephalitis virus and West Nile virus, a number of host factors related to endoplasmic reticulum function and receptors have been screened out. For example, Zhang et al. [7] verified nine human genes required for flavivirus infection through genome-wide screening of CRISPR/Cas9, all of which are related to endoplasmic reticulum (ER) functions (including translocation, protein degradation and N-linked glycosylation). Ma et al. [8] screened the whole genome by designing a library containing 77406 sgRNAs and targeting 20121 genes, and then obtained seven genes belong to the endoplasmic reticulum related protein degradation pathway (ERAD), such as EMC2, EMC3 and SEL1L. Further experiments showed that knockdown of these genes would not prevent the replication of West Nile virus (WNV), but would prevent the virus from killing cells.
It is quite simple to use peptides or purified proteins to verify antibodies in immune experiments, but it is relatively difficult to use the whole cell or other complex antigens to verify antibodies. If the reactivity of the antibody is not detected in Western blotting and immunoprecipitation, it is necessary to apply a variety of techniques at the gene level to determine the antigen specificity of the mAb. The application of CRISPR library can reduce many operation steps and simplify the experiment. BF4 is an antibody that can bind to the viral biofilm on the surface of uninfected lymphocytes, neutrophils and HTLV-1 infected cells. Zotova et al. [9] used MT2 cells (T cells chronically infected with human T-cell lymphotropic virus type I HTLV-1) as immunogens to trigger mouse immunity and obtained a HTLV-1 biofilm specific monoclonal antibody BF4. Based on the idea of screening BF4 antigen knockout cells by transducing CRISPR knockout library into BF4 positive cells, they transduced GeCKO library on CEM T and Raji/CD4 B cells to sort out cells that do not bind to BF4. After two rounds of repeated sorting, the proportion of negative cells reached more than 99%. Researchers sequenced these cells and found that about 80% of sgRNA targeted CD82. BF4 was confirmed to be a specific antibody against CD82.
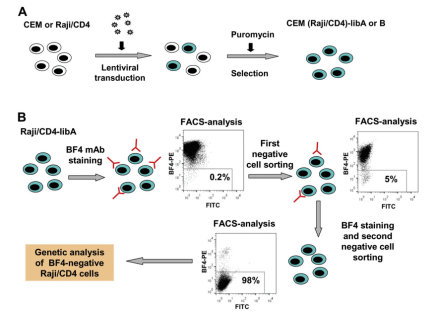
Fig. 4 Verification of BF4 antibody specificity using CRISPR library
Pyroptosis is an immune defense reaction initiated by the body after sensing the infection of pathogenic microorganisms. Inflammation activated caspase-1 and caspase-4, caspase-5 and caspase-11, which recognize bacterial lipopolysaccharides, can cause cell pyroptosis, but the mechanism remains unknown. Shi et al. [10] first established a lipopolysaccharide (LPS) electrotransformation method that can induce more than 90% cell pyroptosis, and then transduced a CRISPR knockout library into Tlr4–/– bone marrow-derived immortalized macrophages (iBMDMs) that can normally respond to lipopolysaccharide stimulation, and sequenced and analyzed the cells that survived after lipopolysaccharide induced cell pyroptosis. The analysis results showed that among the five sgRNAs targeting the gasdermin D (GSDMD) gene, four had copy numbers in the top 30, of which two were in the top 10. The subsequent results also further proved that the N-terminal of GSDMD could induce cell pyroptosis. To sum up, the researchers used CRISPR gene library to carry out genome-wide genetic screening, successfully screened the gene GSDMD that can inhibit cell pyroptosis after knockout, and clarified the molecular mechanism of GSDMD as an inflammatory caspase substrate protein that can trigger cell pyroptosis after being cleaved.
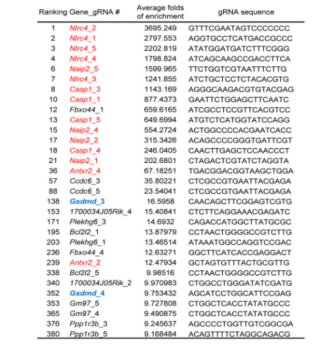
Fig. 5 LFn-Bsak CRISPR-Cas9 screen hits with multiple gRNAs
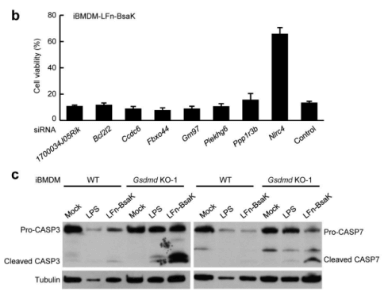
Fig. 6 siRNA knockdown validation of screen hits
According to the size of the library, it can be divided into whole genome library and sub library. The whole genome library usually designs gRNAs for all coding genes of a species, with a large coverage, but its screening workload is huge. Sub library refers to the combination of a certain type of genes, such as a gene family, a signal pathway, etc. At present, there are many ready-made libraries, such as the whole genome library of human, mouse, and green monkey, and sub library of kinase, nuclear protein, cyclin, metabolism related proteins and epigenetic related proteins. Generally, the library coverage should be selected according to the specific needs, followed by the ready-made library, and finally the smallest library that can meet the screening requirements should be selected to minimize the subsequent screening workload.
After determining the screening coverage, we need to consider the screening requirements and clarify whether the purpose of the study is to carry out loss of function screening or gain of function screening? The former generally selects CRISPR-KO or CRISPRi system for construction. Generally speaking, the knockout effect of CRISPR-KO is better, but for cell essential genes (lethal after knockout) or non-coding genes (unable to use frameshift mutation to cause KO), CRISPRi system is a better choice. CRISPRi system is a fusion protein composed of catalytically inactive dCas9 and transcriptional repressor, which is cotransfected with gRNA targeting the upstream regulatory region of the gene to achieve gene knockdown. If gain of function screening is needed, the CRISPRa system is selected. The CRISPRa system enhances gene expression by connecting the catalytically inactive dCas9 with the transcription activator and targeting the upstream regulatory region of the gene.
Other requirements: 1) Barcode library: traditional CRISPR pool screening generally requires phenotypes related to cell proliferation or survival, which is conducive to the enrichment of specific cells. However, if it is an inconspicuous phenotype, such as metabolize, tissue homeostasis, etc., it is necessary to analyze the transcriptome of a single cell with the help of single-cell sequencing technology. In this case, when building the library, it may also be necessary to modify the CRISPR backbone and add the corresponding barcode sequence. 2) Base editing library: use the method of base editing (CBE, ABE, etc.) to build a single base mutation (SNVs) library for a gene or some genes.
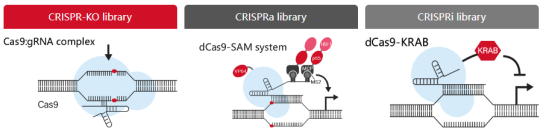
Fig. 7 3different CRISPR libraries
According to different screening purposes, it can be divided into positive screening and negative screening. Positive screening is to exert certain screening pressure on the cells that has successfully integrated sgRNAs, so that only a small number of cells with different phenotypes can survive and enrich key genes. Negative screening is on the contrary. Surviving cells are not target phenotypic cells. It is necessary to compare the abundance of sgRNAs at different time points to find out the difference sgRNAs to determine the key genes. However, negative screening can identify the genes that cause the loss of function of cells. If the screening time is prolonged, genes necessary for cell survival can also be screened. In the application cases mentioned in the previous article, screening driver gene and synthetic lethality gene belongs to negative screening, while screening drug resistance gene and host factor required for viral infection belongs to positive screening.
Design 3-6 sgRNAs for each gene of a species, synthesize sgRNAs using high-throughput chip synthesis method, and then clone the synthesized sgRNAs into lentiviral vector through Gibson method.
Packaging plasmid library into lentivirus, and infecting target cells with low MOI (generally < 0.3), so as to ensure that one virus particle infects one cell. The number of cells contained in the cell library is generally 200-1000 times the number of all sgRNAs.
Divide the whole genome knockout cell bank into two parts, one of which is used as the experimental group to apply screening condition, such as virus infection, drug treatment, etc; The other used as the control group. Cells are screened according to their drug resistance, proliferation ability, viability and other phenotypes.
Genomics are extracted from the cells of the experimental group and the control group, and sgRNA are amplified by PCR, followed by NGS sequencing and bioinformatics analysis.
Knockout a single candidate gene by CRISPR/Cas9, or conduct overexpression or complementation experiments to verify gene function.
Features of Ubigene's CRISPR library service:
①Red Cotton CRISPR gene editing designer:
High throughput automated design of gRNA to ensure gRNA specificity and cutting efficiency.
②Ensure high transformation efficiency:
Use self-developed competent cells, improve transformation efficiency by electro-transformation, and strictly control the number of bacteria ≥ 500X to ensure transformation efficiency. Ensure the library coverage > 99% and uniformity < 10.
③Rich experience in lentivirus packaging:
Ensure the lentivirus titer and purity and the uniformity of the packaging process of mixed plasmid.
④Nearly a hundred Cas9 stable cell lines are available:
Save the time of Cas9 lentivirus infection, and the library screening turnaround is shortened by 3-5 weeks.
⑤Rich experience and sophisticated platform:
Rich experience in cell culture and perfect downstream phenotype analysis platform can provide a variety of cell screening services.
⑥CRISPR-U™ cell line gene editing platform:
It has accumulated more than 5000 successful cases on more than 200 cell lines, provides gene function validation services, and guarantees the delivery of KO homozygous clones.
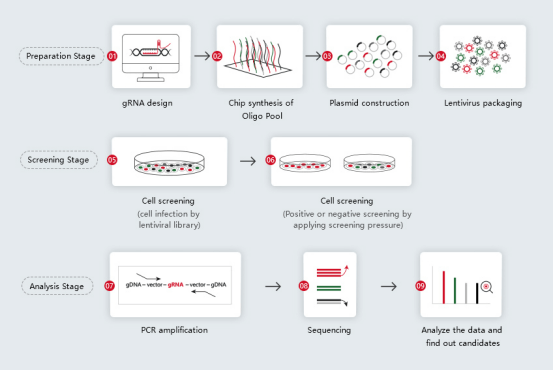
Fig 8. CRISPR library construction and screening process
Reference
[1] Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014 Jan 3;343(6166):84-87.
[2] Wang Y, Gao B, Tan PY, Handoko YA, Sekar K, Deivasigamani A, Seshachalam VP, OuYang HY, Shi M, Xie C, Goh BKP, Ooi LL, Man Hui K. Genome-wide CRISPR knockout screens identify NCAPG as an essential oncogene for hepatocellular carcinoma tumor growth. FASEB J. 2019 Aug;33(8):8759-8770.
[3] Kiessling M K, Schuierer S, Stertz S, et al. Identification of oncogenic driver mutations by genome-wide CRISPR-Cas9 dropout screening[J]. BMC genomics, 2016, 17(1): 1-16.
[4] Sasaki M, Ogiwara H. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex‐deficient cancers[J]. Cancer Science, 2020, 111(3): 774-782.
[5] Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, Sanchez KS, Thomas A, Kuo CC, Du D, Roguev A, Lewis NE, Chang AN, Kreisberg JF, Krogan N, Qi L, Ideker T, Mali P. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods. 2017 Jun;14(6):573-576.
[6] Feng X, Tang M, Dede M, et al. Genome-wide CRISPR screens using isogenic cells reveal vulnerabilities conferred by loss of tumor suppressors[J]. Science advances, 2022, 8(19): eabm6638.
[7] Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016 Jul 7;535(7610):164-8.
[8] Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L, Manjunath N, Wu H. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015 Jul 28;12(4):673-83.
[9] Zotova A, Zotov I, Filatov A, Mazurov D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. J Immunol Methods. 2016 Dec;439:8-14.
[10] Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015 Oct 29;526(7575):660-5.



