Human CRISPR Knockout Pooled Library (Brunello)(Human CRISPR Knockout Pooled Library (Brunello))
Catalog#: LIBR-H015-P002;LIBR-H015-P100;LIBR-H015-P200;LIBR-H015-P500
Size: 2ug
Instruction:

[Useful tips!] How to improve the accuracy of CRISPR library screening targets?

Since the debut of the CRISPR library, it has always been a spotlight in the field of target screening. Many scientific researchers want to use this tool to round off their papers. However, Ubigene always receives some feedback like: Why are the targets I screened not accurate enough?
In fact, the accuracy of library screening will be affected by many factors, and every step should be firmly controlled! Next, Ubigene will introduce several steps that will affect the accuracy of the target in the experiment and the corresponding improvement methods:
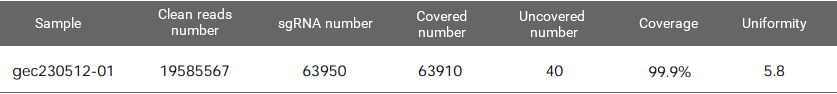
Figure 1. EZ-editor™ Human GeCKO v2 genome-wide library Dual-plasmid System - Library A
Coverage is the number of gRNAs detected by NGS divided by the number of sgRNAs in the theoretical list of the library. The maximum value is 100%. The closer to 100%, the better.
Uniformity refers to the difference in the abundance of different gRNAs. An ideal library has the same abundance for every gRNA, but there will be differences in the gRNA abundance of the actual library. Indicating as the skew ratio, it is the ratio of the number of gRNAs corresponding to the cumulative distribution reaching 90% to the number of gRNAs corresponding to the cumulative distribution reaching 10% (that is, the ratio of 90% quantile and 10% gRNA number). The minimum value is 1, and the closer to 1, the better.
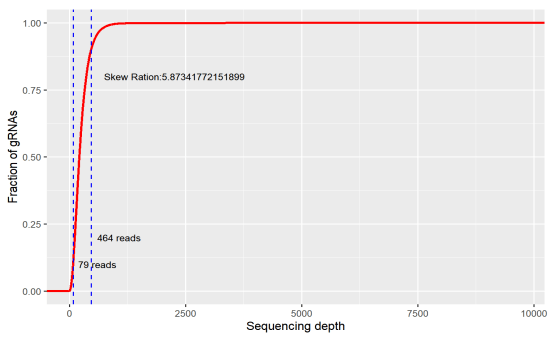
Figure 2. skew ratio
If the coverage of the library is too low, a large number of sgRNA has been lost before the screening. If your ideal target happens to be in it, you will miss it. Or if the uniformity is too poor, a large number of sgRNAs have only very low reads, and the gRNA would be eventually lost during the experiment, which will also affect the final result.
How can we ensure the coverage and uniformity of the plasmid library? At present, most of the commonly used libraries have off-shelf products, therefore customization is not required and only amplification is required. And it must aware in the amplification stage that: 1) select the electroporation method and repeat the electroporation operation several times according to the size of the library; 2) Select ultracompetent cells with transformation efficiency ≥ 109cfu/ug for electroporation 3) Calculate the transformation efficiency, it is required to have the number of colonies ≥ 500 times the number of gRNA.
Different from the virus infection in the conventional construction of stable expression cell lines, the infection efficiency is not the higher the better when infecting the cells by library virus. When there is a large amount of virus, one cell may enter multiple sgRNA, and the final enriched cells are difficult to distinguish which sgRNA plays a role, which may lead to the inaccuracy of the analysis results.
![Different infection efficiencies correspond to the number of sgRNA copies integrated per cell <sup>[1]</sup>](/uploads/allimg/231024/different-infection-efficiencies-correspond-number-sgrna-copies-integrated-per-cell.png)
Figure 3. Different infection efficiencies correspond to the number of sgRNA copies integrated per cell [1]
It can be seen from the figure that the lower the infection efficiency, the less the number of sgRNA copies with two or more in a single cell. However, too low transfection efficiency will in turn require too many cells for infection, so it is necessary to choose a compromise data. The infection efficiency recommended by most literatures is 30%, that is, the Infect MOI is 0.3.
Another concern may come up, that is, how many cells should be used to do the library screening experiment?
For example, if a library virus contains 30000 sgRNAs, according to the infection efficiency requirements in the previous step, only 30% of the cells need to be successfully infected by the virus. Assuming that the number of successfully infected cells is exactly 30000, and the number of uninfected cells is 70000, the total number of cells for infection needs to be 100000. In this extreme case, the number of cells after infection is exactly the same as that of sgRNA, which requires only one virus per cell, which is almost impossible from the perspective of probability. Even if there is only one cell for each sgRNA, it is easy to be lost during passaging. The current guidelines on CRISPR screening recommend that the fold coverage of libraries for cell experiment is between 200X and 1000X, but it lacks an accurate indication of which fold coverage should be used in a given library [2].
The following summarizes the fold coverage used in the library screening-related literature in recent years, including whole genome libraries and sub-libraries, positive screening and negative screening, covering the application of tumor drug resistance research, synthetic lethal gene pairs, viral infection targets and antidote drug development, etc. Most of them choose 500X fold coverage, but also 200X, 400X, etc.
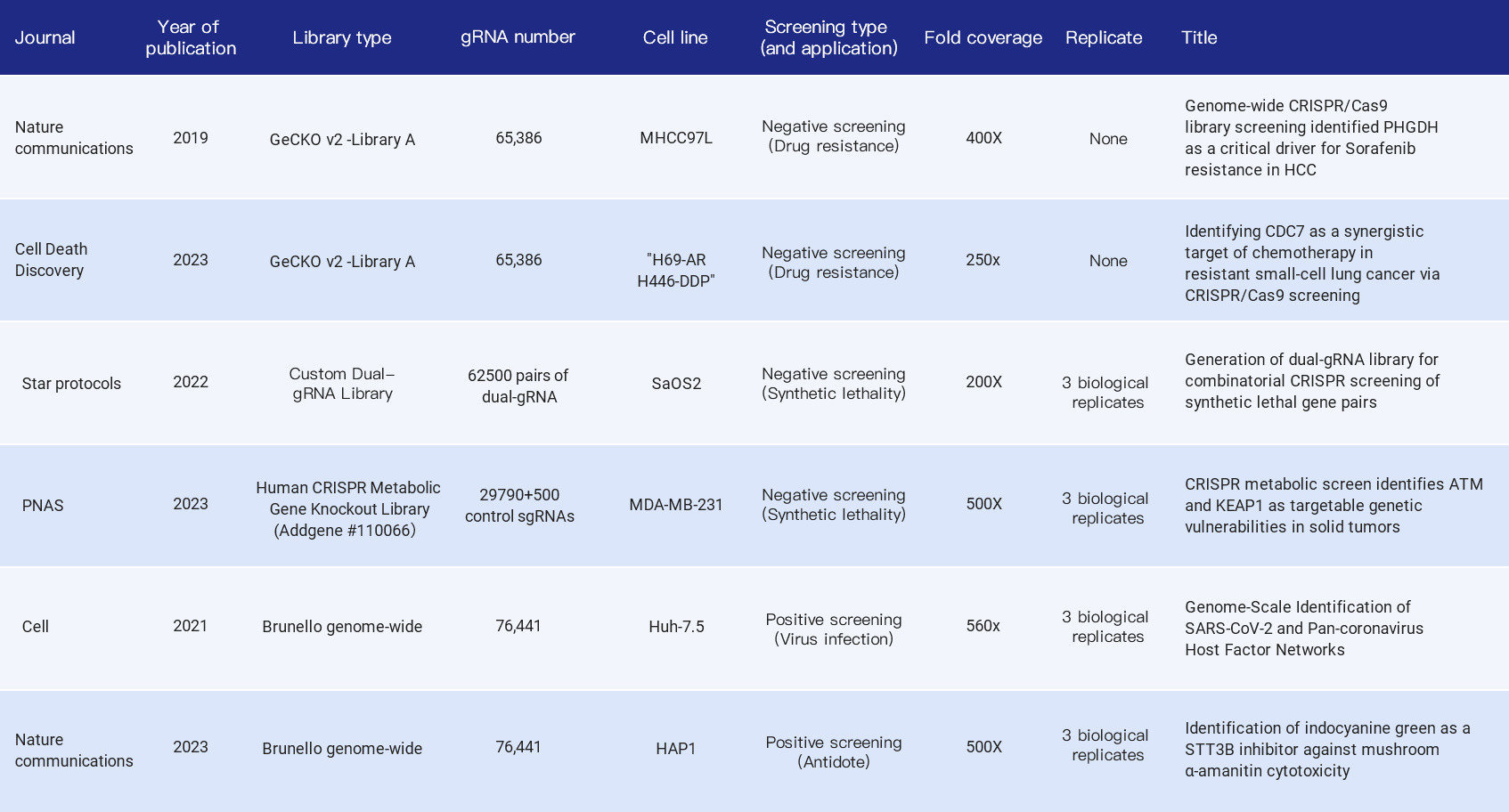
Table 1. Citations indicating fold coverage of CRISPR library in cell screening experiment [3,4,5,6,7,8]
Why do most articles recommend 500X? According to the cell passage model, when the fold coverage is low (such as 50X), it will cause random loss of some sgRNAs per passage, affecting the distribution of the library and the final interpretation of dropout and enrichment. When the fold coverage is >500X, this noise can be greatly reduced (Figure 4). Similarly, the extraction of cell genomes after enrichment and the final NGS sequencing should also ensure the corresponding experimental fold coverage.
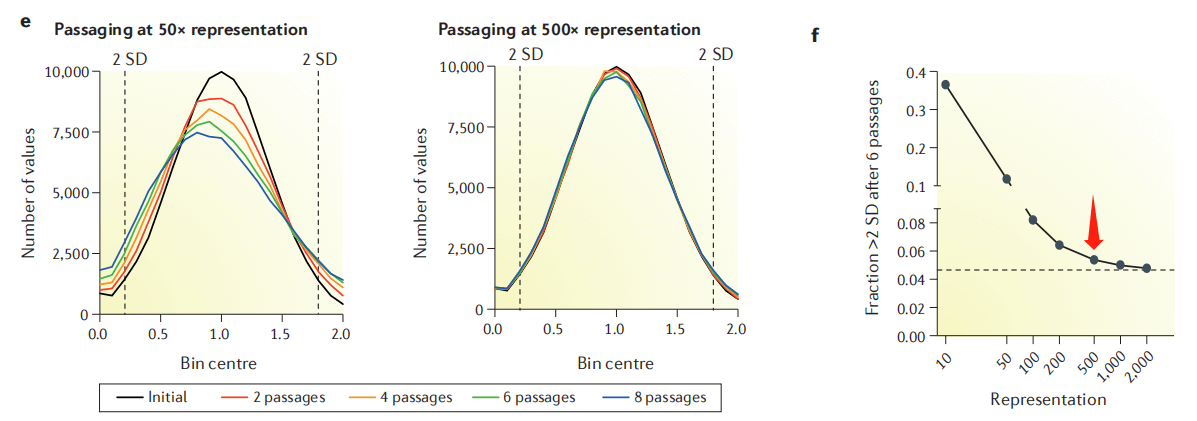
Figure 4. Effect of cell passage coverage on library distribution
Accurate experimental data depends on the firm control of each experimental step. Once a step is not rigorous enough, it may lead to unsatisfactory experimental results, especially the huge library screening experiment.
Conclusion
Optimizing your CRISPR screening library is essential for achieving high-efficiency and reliable gene-editing results. By utilizing advanced strategies for CRISPR screening and carefully designed CRISPR libraries, researchers can maximize target gene coverage, improve screening accuracy, and streamline workflows. Whether you're conducting CRISPR Cas9 screening or exploring a broader CRISPR screen, fine-tuning your approach ensures robust, reproducible outcomes for functional genomics studies. Ubigene's expertise and cutting-edge solutions empower researchers to overcome challenges and achieve breakthroughs in genetic research, driving discoveries and accelerating scientific progress.
References
[1] Doench J G. Am I ready for CRISPR? A user's guide to genetic screens[J]. Nature Reviews Genetics, 2018, 19(2): 67-80.
[2] Diehl V, Wegner M, Grumati P, et al. Minimized combinatorial CRISPR screens identify genetic interactions in autophagy[J]. Nucleic acids research, 2021, 49(10): 5684-5704.
[3] Wei L, Lee D, Law C T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC[J]. Nature communications, 2019, 10(1): 4681.
[4] Deng L, Yang L, Zhu S, et al. Identifying CDC7 as a synergistic target of chemotherapy in resistant small-cell lung cancer via CRISPR/Cas9 screening[J]. Cell Death Discovery, 2023, 9(1): 40.
[5] Tang S, Wu X, Liu J, et al. Generation of dual-gRNA library for combinatorial CRISPR screening of synthetic lethal gene pairs[J]. STAR protocols, 2022, 3(3): 101556.
[6] Li H, Liu Y, Xiao Y, et al. CRISPR metabolic screen identifies ATM and KEAP1 as targetable genetic vulnerabilities in solid tumors[J]. Proceedings of the National Academy of Sciences, 2023, 120(6): e2212072120.
[7] Schneider W M, Luna J M, Hoffmann H H, et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks[J]. Cell, 2021, 184(1): 120-132. e14.
[8] Wang B, Wan A H, Xu Y, et al. Identification of indocyanine green as a STT3B inhibitor against mushroom α-amanitin cytotoxicity[J]. Nature Communications, 2023, 14(1): 2241.



