Gene knockout cell line: How to design gRNAs and avoid protein residues?

Genome editing is a dream method for accurately and precisely recognizing and locating a specific sequence within a genome, and then altering the targeted sequences for various purposes. The CRISPR/Cas9 system has become synonymous with genome editing in today’s world due to its simplistic mechanism. Gene knockout is the most common application of CRISPR/Cas9.
In ‘knockout’ experiments, the intended result is the generation of insertions and deletions (indels) arising from imperfect repair of double-strand DNA breaks by endogenous machinery, an approach that is often used to generate knockout alleles of protein-coding genes or to disrupt transcription factor binding sites. A unifying feature of this experimental interventions is that the Cas enzymes is directed to its genomic destination by a short sequence of homologous RNA, called single guide RNA (sgRNA). There is a couple of considerations that help a researchers to achieved effective gRNA to generate successful knockout. These include predicting gRNA efficiency, minimize gRNA off-target effects, improve knock-out efficiency with design multiple. In a coding gene knockout (KO) experiment, KO result is firstly verified at genome DNA level by genotyping. Moreover, it is critical to validate at protein level, usually by Western blot, which is challenged by the following phenomenons.
(1) Protein expression may persist due to the initiation of translation at alternative downstream ATG codons or simply due to the existence of alternative first coding exons. Translation reinitiation leading to N-terminally truncated target proteins.
(2) Skipping of the edited exon leads to protein isoforms with internal sequence deletions.
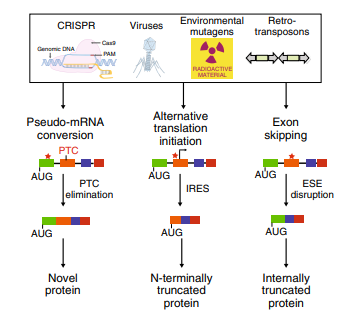
Fig1: Cellular mechanisms for countering INDEL effects revealed by CRISPR failures
Translation reinitiation
The principle of gene knockout with CRISPR technology is usually accomplished by Cas9-mediated dsDNA breaks: following a cut, the error-prone nature of non-homologous end joining (NHEJ) often leads to the generation of indels and thus frameshifts that disrupt the protein-coding capacity of a locus. The cell repair pathway often results in insertion or deletion (indel) of one or more nucleotides at the point of cleavage and generates a nonsense-mediated decay (NMD) that completely knocks out gene function. However, in nonsense mutations near the 5′ region of open reading frames (ORFs), translation can be initiated from an in-frame ATG other than the authentic translation initiation codon. Such illegitimate translation (ITL) proteins have been reported as a genetic factor associated with human diseases. Moreover, ITL has been found in normal genes without mutations; for example, upstream open reading frames (uORFs) regulate gene expression in response to environmental conditions. Thus, out-of-frame mutations established by genome editing may result in protein products due to ITL.
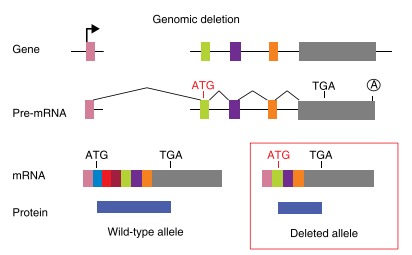
Fig2: Mechanism of Translation reinitiation
Exon skipping
Splicing disruption is a frequent consequence of mutations generated by CRISPR/Cas9 gene-editing technology, and alleles designed to be null can express aberrant proteins. Recently, Mu et al. reported that targeting exons using CRISPR and a single sgRNA in cell lines can produce exon skipping by two mechanisms. The first occurs during splicing of the mutated pre-mRNA, and the second is caused by genomic deletions that remove multiple exons and splicing of the remaining exons.
The mechanism for skipping multiple exons is straightforward: the exons that remain intact after a genomic deletion are spliced. More complex and concerning is the fact that a change of only one to a few nucleotides can result in exon skipping during pre-mRNA splicing. In addition to the splice sites at the intron-exon boundaries, exons contain sequences that act positively or negatively on splicing efficiency. Positive-acting elements within exons, known as exon splicing enhancers (ESEs), bind factors that enhance recognition by the splicing machinery. Negative-acting elements (exon splicing silencers (ESSs) are thought to prevent the use of cryptic splice sites. So, one can hypothesize that an indel promotes exon skipping either by disrupting an ESE or by fortuitously introducing an ESS. Ideally, it would be possible to know a priori how to target a gene and avoid exon-skipping issues. However, it is currently difficult to predict the effect of a given indel on splicing efficiency based on exon sequence. Algorithms to identify exonic splicing elements have been partially successful, and the computational definition of the so-called splicing code is ongoing but these cannot yet completely predict the effect of a given nucleotide change on splicing efficiency.
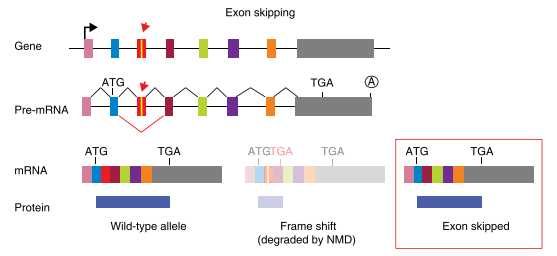
Fig3: Mechanism of exon skipping
What is one to do?
It is important to set up rigorous quality controls to ensure that the mutants created do not contain hidden surprises that could produce artifacts. Ideally, it would be possible to know a priori how to target a gene and avoid exon-skipping issues. Prior to design a KO project one can do the following experiments to avoid potential artifacts of KO mutant validation.
1. It is recommended to use RT-qPCR to determine whether the exon containing skiping capacity, and if so, to determine the protein coding potential. For example, by comparing RT-qPCR results of using primers targeting exons upstream and downstream of the potential skipping exons versus those targeting on the potential exons, one will be able to tell if an exon skipping is happening and whether it could interfere the knockout experiment.
2. Western blot and mass spectrometry are also useful for protein verification before start a KO project. When having a good antibody expected MW band with additional bands at a distinct MW can be present in the WB results, which indicate a presence of protein isoforms.
How to Design your gRNA with confidence?
Earlier, different research groups found that mutation on symmetric exon will highly responsible for plasticity. To avoid plasticity one should design gRNA, which specifically targets asymmetric exon. Additionaly, to avoid pitfall ko validation one should consider some parameters that including gene complete transcript information, conventional and alternative start codons position in the coding region, targeted exon location, exon size, and gRNA score as well as choosing coding-exon based on the number of bases is not multiplied by 3 means asymmetric exon.
In summary, the proper use of CRISPR technology will always depend on careful experimental design, execution, and analysis. The selection of gRNAs for an experiment needs to balance maximizing on-target activity while minimizing off-target activity, which sounds obvious but can often require difficult decisions. In order to obtain KO cell line without protein residue, we must consider all kinds of factors in gRNA design stage. Please note that the high score is not the only standard of choosing a gRNA. The low score gRNA that can target the key site of protein gene is better than the one targeting random position of the gene with the highest score.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!
Ubigene focuses on gene-editing cell line services, and has a gRNA design system - "Red CottonTM CRISPR Gene Editing Designer". In addition, a professional technical team helps researchers design complete gene knockout solutions. We have helped researchers edit genes on more than 100 types of cell lines. Based on experience and data analysis, now we offer a WB guarantee knockout cell line service, 5000 genes are covered in this guarantee.
Reference:
Biological plasticity rescues target activity in CRISPR knockouts. Nature methods (2019), 16(11):1087-1093.
Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell (2014),15:27–30.
Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun (2019), 10(1):5817.
Unexpected consequences: exon skipping caused by CRISPR-generated mutations. Genome Biology (2017) 18:109.
Design and analysis of CRISPR–Cas experiments.Nature biotechnol (2020) 38(7):813-823.



